Les pièges du règlement dispositifs médicaux

Le nouveau règlement 2017/745 commence à être assimilé par les acteurs du dispositif médical et les difficultés se dévoilent à mesure que l’on cherche à construire des réponses, même sur des aspects fondamentaux.
Cet article liste les points durs identifiés, qui feront couler sang, sueur et larmes des RAR et RAQ. La présentation est par ordre d’on est pas sortis de l’auberge : du moins grave au plus bloquant pour le secteur.
Rq : je me permets une proposition pour chaque point, une habitude des commissions de normalisation ;)
Des notices gavées aux avertissements
Le chapitre III de l’annexe I définit les exigences sur les informations à fournir, notamment pour les instructions d’utilisation :
“la notice d’utilisation contient toutes les informations suivantes : […] tout risque résiduel”
Traduction : merci de mettre un message d’avertissement pour chaque ligne de votre analyse des risques, avec à la clé des pages et des pages de warnings qui ne seront probablement jamais lus.
Proposition : confirmer le besoin de fournir une information via la gestion des risques et l’aptitude à l’utilisation.
Des normes volontaires d’application obligatoire
L’annexe VII définit les obligations des organismes notifiés, l’occasion pour les fabricants de comprendre à quelle sauce ils seront mangés :
“l’organisme notifié tient compte des […] normes harmonisées, même si le fabricant ne prétend pas s’y être conformé“
Traduction : ce sont des normes d’applications obligatoires, mais en payantes (c’était déjà le cas avec la directive).
Proposition : un Wikipedia des bonnes pratiques.
De l’amélioration continue
Dans l’article 10 sur les obligations des fabricants :
“les fabricants […] améliorent en permanence un système de gestion de la qualité”
Traduction : c’est le retour de l’amélioration continue, volontairement écartée de l’ISO 13485 car incompatible avec les objectifs de maintient de la conformité des dispositifs (le mieux est l’ennemi du bien).
Proposition : on oublie.
Une définition bancale
Le statut de dispositif médical dépend de la finalité d’utilisation revendiquée par le fabricant, la définition est donnée dans l’article 1 : “destiné au diagnostic, contrôle, .. d’une maladie, d’un handicap…” elle se résume très bien dans un tableau :
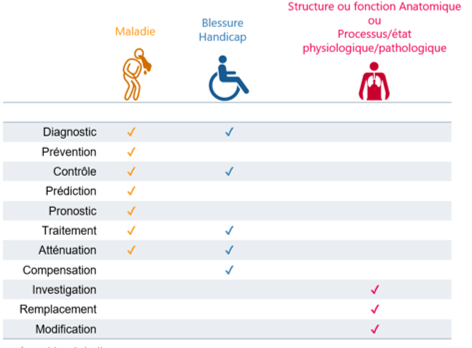
ainsi, un produit destiné à :
- prédire l’évolution d’un handicap
- compenser les effets d’une maladie
- prédire un état physiologique
- contrôler un état pathologique
- …
n’est pas DM.
Proposition : “un dispositif médical est un produit pour lequel le fabricant revendique un bénéfice clinique“.
(La notion de bénéfice est développée dans le règlement, avec 4 types de bénéfices: résultat thérapeutique, résultat diagnostic, amélioration de la prise en charge du patient ou amélioration de la santé publique).
Des investigations cliniques systématiques
Classe III et implantable sont gâtés : l’investigation clinique est obligatoire, merci l’article 61 :
“Dans le cas des dispositifs implantables et des dispositifs de classe III, des investigations cliniques sont conduites“
Traduction : la nécessité de recourir à une investigation n’est pas liée aux données cliniques disponibles.
Un choix aussi binaire que discutable, il y a des classes I qui ont beaucoup plus besoin de “data fraiche” que certains DM implantables déjà ultras maitrisés.
Attendez vous à une saturation du marché de l’investigation clinique entre 2019 et 2025 (en 2026 tout le monde aura abandonné).
Proposition : pour les classes III et implantable, l’ON évalue le besoin de lancer une investigation, en tenant compte des données cliniques présentées par le fabricant, et si besoin en sollicitant un groupe d’expert européen et Michel Cymes.
Une classification à la truelle
Voir l’article sur les changements des règles de classification du règlement DM.
Rappel : l’idée est de classer un dispositif selon sa dangerosité potentielle, une démarche assez subtile qui nécessite tout un tas d’activité de gestion des risques, malheureusement réduite ici à 80 critères techniques qu’il faudra sans cesse consolider, au fil des cas borderline.
Certains critères sont discutables, avec des choix malheureux repris de la 93/42 :
- Règle 5 : le fabricant d’un DM invasif utilisé 59 minutes sera en auto-certification (classe I), 1 minute de plus et c’était 2 ans et 100k€ à ajouter pour le CE (classe IIa).
- Règle 8 : les DM implantables relèvent de la classe IIb avec des cas particuliers en classe III comme pour les implants
PIPmammaires; les implants testiculaires attendront leur scandale sanitaire. - Règle 9 : un DM actif thérapeutique fournissant de l’énergie est classé IIa sauf s’il est (potentiellement) dangereux auquel cas c’est IIb. Je reformule : la classe de dangerosité d’un DM non dangereux fournissant de l’énergie est IIa. Signé : le front de lutte contre la classe I. C’est le même problème avec la Règle 10.
- Règle 11 : un logiciel fournissant une information à des fins thérapeutiques ou diagnostics est classé en fonction de la sévérité des risques associés : III si mort ou détérioration irréversible, IIb si grave détérioration ou intervention chirurgicale, IIa sinon.
Et c’est tout, pas de nuance type “IIa si dommage non grave et I si pas de dommage” : la classe I n’existe pas.
C’est pire pour les logiciels destinés à contrôler un processus physiologique qui oublient les classes III et I : IIb si processus vital et danger immédiat, IIa sinon.
Pour finir de pleurer : notez que la règle pour le logiciel est désynchronisée de la définition de DM :
- “contrôler un processus physiologique” n’est pas inclus dans la définition
- tout ce qui est prévention, prédiction, pronostic, atténuation, compensation, investigation, … n’est pas pris en compte => classe I par défaut.
- Règle 18 : vous utilisez des nano matériau ? IIa même si le patient n’est pas exposé.
- Règle 20 : vous administrez un médicament ? IIb max. S’il y a un mort on dira que c’est la faute à la règlementation pharma.
Proposition : définir la classe de risque … en fonction des risques : III si risque de mort; IIb si blessure irréversible, IIa si réversible, I si simple gène.. vous voyez l’idée.
Une maitrise des risques ad-libitum
Apporter des bénéfices cliniques et gérer les risques : c’est le nerf de la guerre pour un fabricant de DM. Le règlement développe des exigences en matière de gestion des risques dans l’annexe I.I (exigences générales) qui précise quand arrêter la maitrise des risques :
“L’exigence de la présente annexe prévoyant qu’il convient de réduire les risques autant que possible signifie réduire les risques autant que possible sans altérer le rapport bénéfice/risque“
Traduction : pour chaque risque identifié vous allez truffer le dispositif de composants haute fiabilité, protections, redondance, marquages,… avec des kilos de pages de warnings et une caméra pour qu’un technicien puisse vérifier la bonne utilisation du DM.
Notez qu’un DM parfaitement sécurisé sera hors de prix => pas de vente => pas d’incident => rapport bénéfice / risque favorable.
Proposition : la réduction des risques s’arrêtent lorsque le rapport B/R est jugé favorable selon la politique définie par le fabricant.
Des investigations cliniques systématiques (bis)
Surement LE point dur du règlement, article 61.5 :
“Le fabricant d’un dispositif pour lequel il a été démontré qu’il est équivalent à un dispositif déjà commercialisé et non fabriqué par lui, peut ne pas conduire d’investigation clinique pour autant que les conditions suivantes soient remplies (en plus du .4):
– les deux fabricants ont conclu un contrat qui accorde explicitement au fabricant du second dispositif un accès total et permanent à la DT, et
– l’évaluation clinique d’origine a été effectuée conformément aux exigences du présent règlement,et le fabricant du second dispositif en apporte la preuve manifeste à l’organisme notifié.”
Ainsi, vous coupez à l’investigation clinique si votre concurrent vous refile son dossier de certification.
Proposition : espérer que cela soit mal formulé et que cela concerne uniquement les classes III et implantables
Conclusion
Respecter la règlementation est long et cher, il faudrait au moins que cela soit cohérent. Espérons que ces points bloquants ne seront pas appliqués de manière trop littérale, pour éviter un fiasco type annexe Z de l’ISO 14971.
Heureusement, une parade a été imaginée par l’Europe : avec plus du quart des organismes notifiés qui abandonnent le médical les fabricants ne devraient même pas avoir l’occasion de se frotter au règlement !
10 commentaires