Directive 93/42/CEE relative aux Dispositifs Médicaux

La directive Européenne 93/42/CEE relative aux dispositifs médicaux (DM) est applicable par tous les États membres de l’union, elle précise les rôles et obligations des différents acteurs du DM.
Cet article, à destination des fabricants, présente les principaux points de la directive.
👉 Attention, la directive a été abrogée, c’est maintenant le règlement (ue) 2017/745 qui s’applique en matière de dispositifs médicaux
Le texte définissant la réglementation Européenne en matière de dispositifs médicaux, assurant la libre circulation de dispositif sûrs et efficaces.
Comme les autres États membres la France transcrit les exigences de la directive dans son droit national, mais elle est maintenant remplacée par le règlement (UE) 2017/745.
La dernière version consolidée est disponible au format pdf.
Principalement : des exigences et des procédures d’évaluation de la conformité. Également des définitions et des explications de notions comme “normes harmonisées”, “organisme notifié”, “classe d’un DM”…
Présentation de la directive 93/42/CEE
Le titre du document renseigne très bien sa nature :
Directive 93/42/CEE du conseil du 14 juin 1993 relative aux dispositifs médicaux
Il s’agit d’une directive Européenne : un texte réglementaire rédigé par le conseil de l’Union Européenne (UE) qui définit des objectifs communs pour les États membres, ces États transcrivent les exigences dans leur droit national, c’est le cas de la France dans des arrêtés et autres décisions regroupés sur cette page.
Initialement publiée en 1993 (42 étant un numéro incrémental) elle a été modifiée en 1998, 2000, 2001, 2003 et finalement par la directive 2007/47/CE de 2007.
La dernière mise à jour est “la version M5” : la 93/42/CEE modifiée par les publications ultérieures, ce document est donc suffisant pour tenir compte des évolutions réglementaires : on parle de version consolidée.
►M5 texte issu de la 5ᵉ Modification◄
La directive concerne les dispositifs médicaux, la finalité du texte est de garantir les performances et la sécurité des produits mis sur le marché Européen tout en proposant un socle commun en adéquation avec la libre circulation des marchandises.
Le respect des exigences de la directive est matérialisé par le marquage CE, valable pour les dispositifs fabriqués en Europe et partout ailleurs dans le monde.
Les principaux articles de la directive
La 93/42/CEE comporte 23 articles, comptez en fait 27 articles : certains sont déclinés en versions bis et ter, mais soyez rassuré : un fabricant de dispositifs médicaux n’a pas à les connaitre par cœur, il suffit de comprendre le contenu des principaux articles listés ci-dessous.
Article 1 : “Définitions, champ d’application”
Outre la définition de dispositif médical, on y trouve les définitions des principales catégories de DM, celles de “mandataire”, de “données cliniques”, de “mise sur le marché”…
Cet article est très important : l’applicabilité des exigences de la directive se base sur ces définitions.
Voir en complément le guide MEDDEV 2.1/1 qui précise les définitions de “DM”, “accessoire” et “fabricant”.
Article 3 : “Exigences essentielles”
Les dispositifs médicaux doivent répondre aux exigences essentielles de la directive, elles concernent les performances du produit, la documentation fournie, les processus à mettre en place par le fabricant… Ces exigences sont détaillées en annexe I.
Article 5 : “Renvoi aux normes”
La mise en œuvre des normes harmonisées est un moyen reconnu pour répondre aux exigences de la directive. Ces normes sont très diverses : gestion des risques avec l’ISO 14971, sécurité des dispositifs électromédicaux avec l’IEC 60601-1, système de management de la qualité avec l’ISO 13485:2016, …
La liste des normes harmonisées à la 93/42/CEE est régulièrement publiée dans le journal officiel de la communauté Européenne.
Article 9 : “Classification”
La classification des dispositifs médicaux se fait selon 4 classes : I, IIa, IIb et III par ordre de dangerosité potentielle croissante.
Les règles de classification sont données dans l’annexe IX.
Cette annexe est relativement complexe, je vous propose d’utiliser ce formulaire de calcul de la classe d’un DM.
De la classe du dispositif dépendent les modalités de vérification CE et les obligations du fabricant envers les autorités (en France les fabricants doivent déclarer les DM à partir de la classe IIa, la liste des DM déclarés est – trop rarement – publiée par l’ANSM).
Article 10 : “Informations sur des incidents intervenus après la mise des dispositifs sur le marché”
Les incidents graves (mort ou dégradation sévère de la santé) doivent être déclarés à l’autorité compétente (l’ANSM en France, voir la page de déclaration d’effet indésirable).
Le fabricant est sollicité pour évaluer les conséquences et les suites à donner, ceci étant partie intégrante du processus de gestion des risques et viendra alimenter le dossier de gestion des risques qui suit la vie du produit.
De tels incidents peuvent être dus à une mauvaise utilisation, un défaut d’information, un dysfonctionnement du dispositif… notez que les causes sont souvent multiples.
Article 11 : “Évaluation de la conformité”
L’article 11 définit les procédures d’évaluation de la conformité des produits : mise en place d’une assurance qualité, intervention d’un organisme notifié… Tout dépend de la classe du dispositif et des choix “organisationnels” du fabricant.
L’article résumé dans cette illustration :
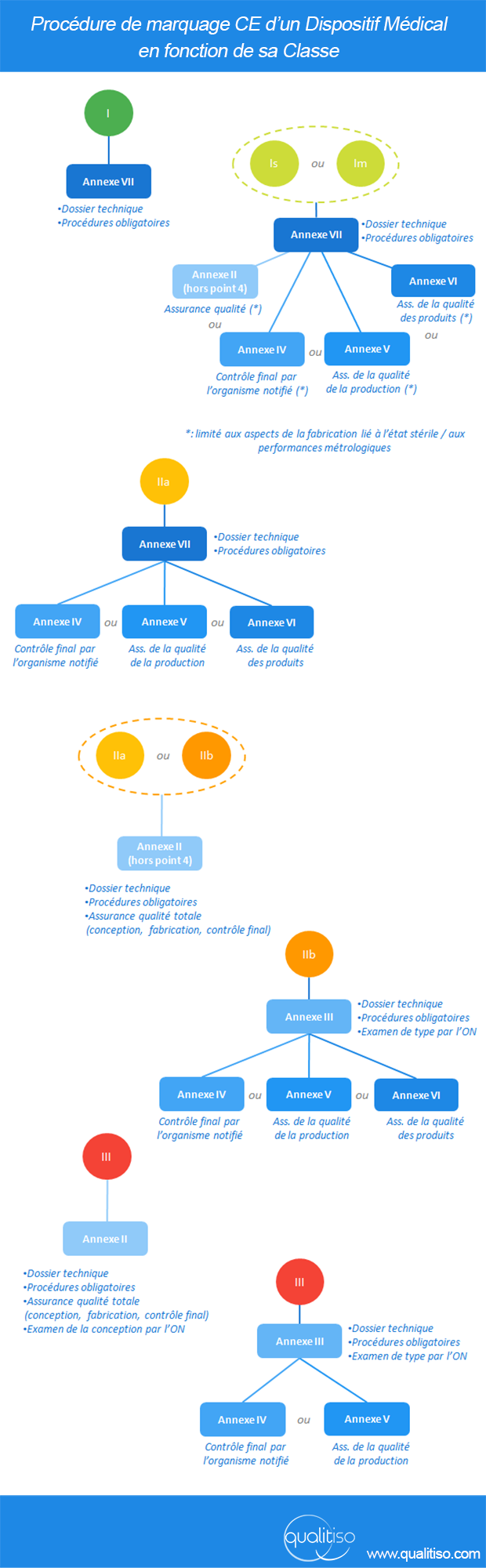
cliquez pour agrandir
Article 14 : “Enregistrement des personnes responsables pour la mise des dispositifs sur le marché”
Le fabricant déclare à l’autorité nationale ses activités et la mise sur le marché des DM (pour les classes supérieures à la classe I).
Un fabricant hors UE doit désigner un mandataire.
Article 15 : “Investigations cliniques”
Dresse les spécificités du marquage CE des DM destinés à des investigations cliniques (procédure définie en annexe VIII).
Les investigations cliniques sont définies en annexe X.
Article 16 : “Organismes notifiés”
Les organismes notifiés (ON) sont des organismes qui, en fonction de la procédure de marquage CE, sont susceptibles d’approuver votre assurance qualité, de réaliser un examen de type, d’effectuer le contrôle final des produits…
Une liste des organismes notifiés est tenue à jour sur le site de la commission Européenne.
Les annexes de la 93/42/CEE
Les annexes constituent la partie la plus “opérationnelle” de la directive:
- Annexe I: les exigences essentielles. Il faudra déterminer l’applicabilité et les moyens de mise en conformité en vue d’obtenir le marquage CE.
- Annexe II à VII: les différentes procédures de marquage CE, en fonction de la classe du DM.
- Annexe VIII: déclaration CE pour les dispositifs “spéciaux”: les dispositifs sur mesure ou destinés aux investigations cliniques.
- Annexe IX: permet de déterminer la classe d’un DM, avec des définitions, des règles d’applications et des règles de classification.
- Annexe X: concerne l’évaluation clinique.
- Annexe XI: concerne la désignation des ON.
- Annexe XII: l’art et la manière de faire un joli marquage CE, pas de modèle vectoriel, mais un bon vieux dessin dans une grille :

5 commentaires