Post-marketing surveillance (PMS) for medical devices
Post-marketing surveillance (PMS) for medical devices (MD) is a major development of Regulation (EU) 2017/745 which, beyond incident management, and requires you to proactively monitor the benefit/risk ratio of your product, during its entire life cycle. The importance of Post-marketing surveillance has significantly increased under the Medical Devices Regulation (MDR).
This article provides an overview of the general PMS requirements, phases and format, taking into account the regulation, the MDCG (Medical Device Coordination Group) guides and the applicable standards.
Requirements for Post-marketing surveillance
The context is set by Regulation 2017/745:
- Article 83: PMS system implemented by the manufacturer (note that PMS also involves distributors and importers);
- article 84: PMS plan;
- Article 85: PMS report;
- Article 86: PSUR;
- Article 88: Trend report; and
- Annex III: PMS Technical Documentation.
These activities will be associated with the processes already in place according to :
- ISO 13485 : with its requirements for measurement, communication, CAPA…
- Guide ISO/TR 20416 with PMS techniques for Software Item manufacturers.
- ISO 14971: and its post-production activities (§9). In general, risk management is very much in demand: from the definition of indicators to data analysis.
- XP S99-223: management of the benefit-risk ratio, defining PMS needs according to identified key data, risks and benefits.
- ISO 14155: for clinical investigation and PMCF activities (clinical surveillance, to be detailed in a dedicated procedure).
- IEC 62366-1: to define and monitor conditions of use
As well as more technical standards such as the IEC 60601-1 series which will be useful for you to identify security and performance related features: all potential indicators for PMS.
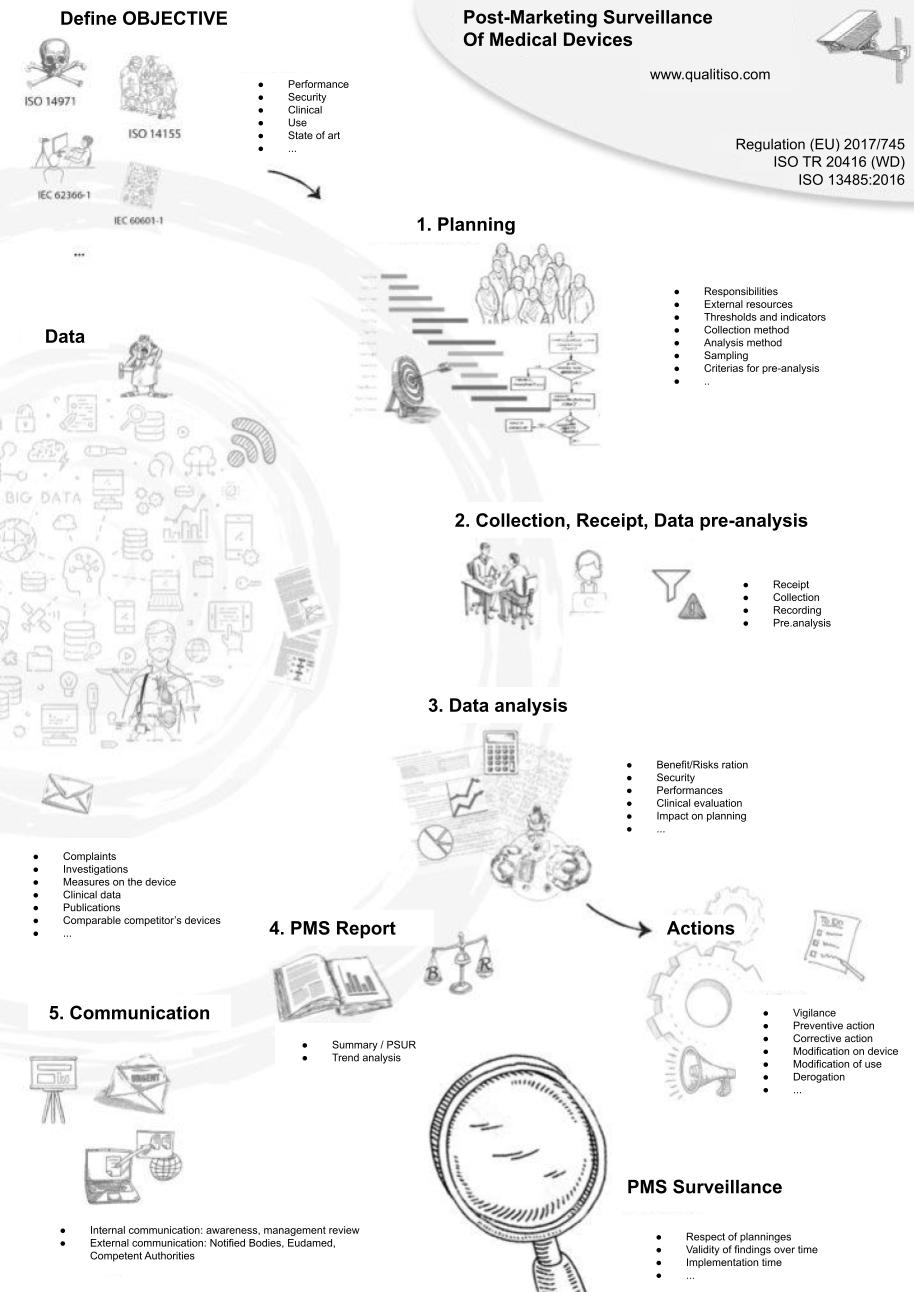
Defining Post-marketing surveillance objectives
Surveillance objectives are identified at the design stage :
- Confirm security and Performance defined during design and development
- Confirm estimates of Key Risks and Benefits, managing the B/R ratio
- Obtain new clinical data to address uncertainties identified during the clinical evaluation (the PMS then becomes the PMCF).
- Consider the state of the art that feed the technical file
Surveillance planning
Planning is built around the objectives, it covers:
- The indicators and thresholds associated with the objectives
- The data targeted: nature, sampling, frequency…
- The methods of collection and reception
- The criteria for pre-analysis of the raw data (and training to those who will receive the information from users)
- The methods of data analysis
- The responsibilities and resources required
- The information expected in the summaries
- The communication needs of the results: internally and to other organisations
A potentially very substantial piece of work … a proportionate approach to the risks will be needed to define a necessary and sufficient scope in relation to the objectives.
Data collection, receipt and pre-analysis
Note the collection/receipt distinction, to emphasise the proactive nature that the PMS must have: Post-marketing surveillance is an active process.
- Collection: measurement on the device, user surveys, technical and clinical watch, surveillance of competing devices…
- Reception: management of complaints and other spontaneous feedback
- Pre-analysis : a quick analysis when receiving raw data (e.g. a phone call from a user) should allow to detect relevant information for the PMS, so that the information is immediately “referred” to the relevant actors.
Analysis of surveillance data
To identify an impact on the evaluation of the benefit/risk ratio or any other relevant information on the product, its use and the medical context surrounding it.
The results and conclusions of the analysis are recorded.
Actions decided following analysis
Exit from surveillance to initiate other processes:
- Corrective and preventive action management
- communication with customers
- change to the device
- change in the intended use
And for the most critical cases: vigilance actions.
Reports and summaries
Top management, your staff, your Notified Body, your Competent Authority… are not going to analyse your raw data or look at your analysis reports, they are working on summaries produced by the manufacturers.
The regulation requires:
- a simple summary for classes I, to be kept in one’s documentation
- a periodic summary (PSUR) from class IIa onwards, to be published on Eudamed for class III and implantable, in any case it will be very much observed by your Notified Body.
The content of the report should provide an understanding of the PMS performed and the highlights. It will state:
- Scope of the PMS
- Summary of findings and conclusions, including benefit/risk ratio
- Summary of actions elicited by PMS
- Estimation of use of the device: number of users, frequency…
Trend analysis
This is quite new and deserves some clarification:
- trend analysis requires a consistent amount of data
- the analysis is “graphic” in nature: representations over time and other distribution diagrams allow the detection of :
- a periodic pattern (e.g. a failure every 100 uses)
- an event correlated to a time of year (failures during hot periods)
- a growth trend
- a trend of acceleration
- …
Depending on the data available and the level of risk the trend analysis will be very simple or very complicated, but it is a powerful tool: the maths is as relevant for making capital gains as it is for reducing the morbidity rate associated with the use of a Medical Devices.
Communication of results
Your Notified Body, Eudamed and your Competent Authority will be interested in your reports.
The PMS results will also be communicated internally, at a management review or to raise awareness among employees.
Monitoring surveillance
Some tips for monitoring your surveillance:
- measure the gap between your planning and your actual PMS
- measure processing times (receipt, analysis, implementation of actions)
- evaluate the sustainability of findings over time
- perform a causal analysis in case of missed information detected after the fact
- …
Flowchart: Post-marketing surveillance phases