Fiche mémo — Investigations Cliniques selon le MDR
Cette fiche fait une cartographie des exigences du règlement (UE) 2017/745 en matière d’investigations cliniques.
Plusieurs éléments sont à considérer: dispositif CE ou non, classe de risque, étude dans le cadre de l’évaluation de la conformité ou non, utilisation du dispositif dans sa destination prévue ou au delà, étude de suivi (PMCF) avec ou sans procédures additionnelles.
Un arbre de décision vous permet de détecter les articles applicables pour vos investigations.
Un résumé des articles du MDR relatif aux investigations cliniques vous est également proposé.
Voir également les exigences nationales des États Membres pour les investigations cliniques.
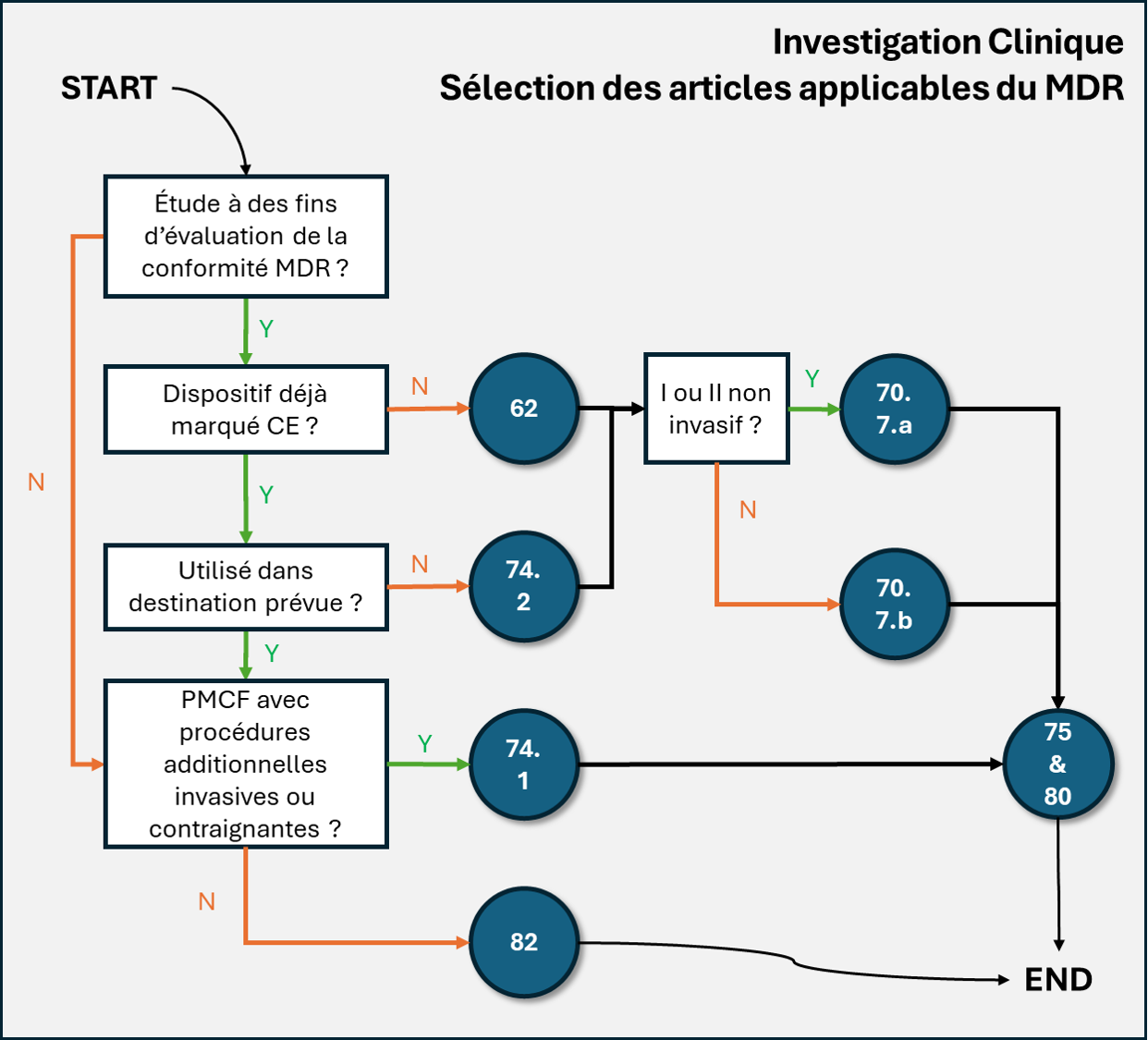
Arbre de décision
Étape 1 — L’étude est-elle menée à des fins d’évaluation de la conformité MDR ?
- Oui → aller à l’étape 2
- Non → aller à l’étape 5
Étape 2 — Le dispositif est-il déjà marqué CE ?
- Non → article 62
- puis appliquer article 70 selon la classe du dispositif
- classe I ou classe IIa/IIb non invasif → article 70(7)(a)
- classe IIa/IIb invasif ou classe III → article 70(7)(b)
- pendant l’étude, appliquer aussi si nécessaire article 75 et article 80
- vérifier en parallèle les exigences nationales des États membres
- Oui → aller à l’étape 3
Étape 3 — Le dispositif marqué CE est-il utilisé dans sa destination prévue ?
- Non → article 74(2)
- les articles 62 à 81 s’appliquent
- en pratique, appliquer aussi la logique de l’article 70 selon la classe du dispositif
- classe I ou classe IIa/IIb non invasif → article 70(7)(a)
- classe IIa/IIb invasif, ou classe III → article 70(7)(b)
- appliquer si nécessaire article 75 et article 80
- vérifier les exigences nationales des États membres, y compris autorité compétente et comité d’éthique
- Oui → aller à l’étape 4
Étape 4 — S’agit-il d’une PMCF avec procédures additionnelles invasives ou contraignantes ?
- Oui → article 74(1)
- appliquer aussi article 75 si modification substantielle
- appliquer les dispositions pertinentes de l’article 80 pour la sécurité
- tenir compte des exigences des États membres, notamment notification, autorité compétente, comité d’éthique, langue et délais
- Non → article 82
- vérifier les exigences nationales applicables dans chaque État membre
Étape 5 — Si l’étude n’est pas menée à des fins de conformité, relève-t-elle de l’article 74(1) ?
- Oui → article 74(1)
- appliquer aussi article 75 si modification substantielle
- appliquer les dispositions pertinentes de l’article 80 pour la sécurité
- tenir compte des exigences des États membres
- Non → article 82
- vérifier les exigences nationales applicables dans chaque État membre
Explication des articles du MDR relatifs aux investigations cliniques
Article 62 — Règles générales des investigations cliniques de conformité
Article de base quand l’investigation clinique sert à démontrer la conformité du dispositif au MDR. Il impose l’autorisation, la revue éthique, la protection des sujets, le consentement, la qualification des investigateurs et le respect de l’annexe XV.
Les États membres peuvent ajouter/préciser : procédure du comité d’éthique, portée de son avis, qualification reconnue de l’investigateur, organisation des soins aux sujets, exigence d’un représentant/contact local dans certains cas.
Article 63 — Consentement éclairé
Fixe les règles de forme et de contenu du consentement : information claire, entretien préalable, signature, droit de retrait, information sur l’indemnisation et sur les résultats de l’étude.
Les États membres peuvent ajouter/préciser : langue des documents, personne habilitée à mener l’entretien, formalités complémentaires de consentement, règles d’assentiment.
Article 64 — Sujets incapables
Conditions supplémentaires si l’étude inclut des personnes incapables de consentir : représentant légalement désigné, information adaptée, bénéfice direct attendu, nécessité scientifique.
Les États membres peuvent ajouter/préciser : définition et désignation du représentant, protections supplémentaires.
Article 65 — Mineurs
Conditions spécifiques pour inclure des mineurs : consentement du représentant, information adaptée à l’âge, respect du refus du mineur, assentiment si possible, nouveau consentement à la majorité.
Les États membres peuvent ajouter/préciser : règles d’assentiment, âge de majorité, représentant légal, protections renforcées.
Article 66 — Femmes enceintes ou allaitantes
Étude possible seulement si le bénéfice direct attendu justifie les risques, avec précautions particulières.
Les États membres peuvent ajouter/préciser : exigences éthiques ou protections complémentaires via leur droit national.
Article 67 — Mesures nationales additionnelles
Article important car il autorise explicitement les États membres à maintenir des règles supplémentaires pour certaines populations vulnérables.
Exemples : personnes privées de liberté, service militaire obligatoire, personnes sous décision judiciaire, personnes en institution.
Article 68 — Situations d’urgence
Permet, sous conditions strictes, une inclusion avant consentement préalable, avec consentement différé ensuite.
Les États membres peuvent ajouter/préciser : modalités pratiques d’acceptation, circuit éthique, représentant légal, étapes procédurales locales.
Article 69 — Indemnisation des dommages
Chaque État membre doit prévoir un système d’indemnisation des dommages subis par les sujets. En pratique, le promoteur doit disposer de l’assurance, garantie ou mécanisme équivalent requis.
Les États membres peuvent ajouter/préciser : type de couverture, niveau de garantie, justificatifs à fournir, modalités locales.
Article 70 — Demande d’investigation clinique
Décrit la soumission du dossier, la validation, la complétude, les demandes d’informations complémentaires et les délais.
- 70(7)(a) : démarrage après validation pour les dispositifs investigués de classe I et les dispositifs non invasifs de classe IIa ou IIb, sauf si le droit national prévoit autrement, et sous réserve de l’avis éthique
- 70(7)(b) : autorisation explicite requise pour les dispositifs non couverts par 70(7)(a)
Les États membres peuvent ajouter/préciser : exigences de langue, pièces administratives, modalités pratiques de soumission, interaction avec le comité d’éthique, et, pour 70(7)(a), imposer un régime plus encadré.
Article 71 — Évaluation par les États membres
Organise l’examen du dossier par l’État membre concerné.
Les États membres peuvent ajouter/préciser : répartition des rôles entre autorité compétente et comité d’éthique, experts consultés, organisation interne de l’évaluation.
Article 72 — Conduite de l’investigation clinique
Couvre la conduite pratique de l’étude selon le plan, le respect continu des conditions de sécurité et la gestion réglementaire en cours d’étude.
Les États membres peuvent ajouter/préciser : modalités de contrôle, échanges avec l’autorité, mise en œuvre pratique de certaines obligations.
Article 73 — Système électronique
Prévoit le système électronique européen pour les soumissions et échanges réglementaires.
Les États membres peuvent ajouter/préciser : quelques formalités nationales complémentaires en pratique, si leur organisation interne l’exige.
Article 74 — Investigations sur dispositifs déjà marqués CE
- 74(1) : étude PMCF sur un dispositif déjà marqué CE, utilisé dans sa destination prévue, avec procédures additionnelles invasives ou contraignantes
- 74(2) : dispositif marqué CE utilisé hors destination prévue ; dans ce cas, les articles 62 à 81 s’appliquent
Jargon :
- PMCF = suivi clinique après commercialisation
- Destination prévue = usage revendiqué par le fabricant
- Procédure additionnelle invasive ou contraignante = acte supplémentaire imposé par le protocole et non requis par la pratique clinique normale
Les États membres peuvent ajouter/préciser : modalités de notification, rôle du comité d’éthique, documents locaux, délais pratiques.
Article 75 — Modifications substantielles
Toute modification importante pour la sécurité, les droits des sujets ou la fiabilité des données doit être notifiée avant mise en œuvre.
Les États membres peuvent ajouter/préciser : procédure, documents, délais, articulation avec le comité d’éthique.
Article 76 — Mesures correctives et échanges d’information
Permet aux autorités de réagir si l’étude présente un risque ou n’est pas conforme.
Les États membres peuvent ajouter/préciser : circuits de notification, autorités destinataires, mesures pratiques demandées.
Article 77 — Fin, suspension, arrêt anticipé et résultats
Le promoteur doit notifier la fin de l’étude, une suspension ou un arrêt anticipé, puis soumettre le rapport final et son résumé.
Les États membres peuvent ajouter/préciser : modalités de notification, langue, attentes locales du comité d’éthique ou de l’autorité compétente.
Article 78 — Procédure coordonnée d’évaluation
Prévoit une évaluation coordonnée entre plusieurs États membres pour certaines études multicentriques.
Les États membres peuvent ajouter/préciser : leur participation et leurs volets nationaux, surtout éthiques.
Article 79 — Réexamen / recours procédural
Article surtout procédural sur le fonctionnement du dispositif coordonné et des suites administratives.
Les États membres peuvent ajouter/préciser : voies de recours, délais et autorités compétentes selon leur droit national.
Article 80 — Enregistrement et notification des événements indésirables
Article clé de vigilance pendant l’étude : événements indésirables graves, déficiences du dispositif, mesures correctives de sécurité.
Les États membres peuvent ajouter/préciser : circuits pratiques de notification, destinataires nationaux et exigences opérationnelles complémentaires.
Article 81 — Actes d’exécution
Permet à la Commission de préciser certains aspects pratiques pour une application uniforme.
En pratique : peu d’impact direct immédiat pour le fabricant au niveau de l’article lui-même.
Article 82 — Autres investigations cliniques
Concerne les investigations qui ne relèvent ni de l’article 62(1) ni de l’article 74(1). C’est la zone où les exigences nationales sont les plus importantes.
Les États membres peuvent ajouter/préciser largement : autorisation ou notification, autorité compétente, comité d’éthique, documents, délais, langue, assurance, représentant local et autres formalités.
Bonus : Annexe XV — Cadre opérationnel des investigations cliniques
Elle précise surtout :
- les documents à soumettre
- le contenu du plan d’investigation clinique
- le contenu du rapport d’investigation clinique
En pratique, dès qu’une étude relève de l’article 62 ou de l’article 74(1), l’annexe XV est centrale.