IAF MD 9:2022 : appliquer l’ISO/IEC 17021-1 pour les audits ISO 13485
L’IAF (International Accreditation Forum) a publié un document, à destination des organismes d’accréditation, détaillant la mise en œuvre de l’ISO/IEC 17021-1 (Évaluation de la conformité — Exigences pour les organismes procédant à l’audit et à la certification des systèmes de management) pour des audits selon l’ISO 13485.
{kind=link}
Attention, ce document ne tient pas compte du contexte réglementaire, uniquement des normes.
Maintien de la conformité
Le document rappelle les rôles de l’organisme notifié et du fabricant:
- L’ON est chargé de déterminer que le fabricant a évalué la conformité légale et réglementaire et peut démontrer que des mesures appropriées ont été prises en cas de non-conformité avec la législation et la réglementation pertinentes.
- Le maintien et l’évaluation de la conformité légale relèvent de la responsabilité du fabricant.
Exemples de non-conformités majeures
Le document détaille la notion de NC majeure, avec des exemples :
- Incapacité à répondre pleinement aux exigences applicables et à mettre en œuvre un processus complet pour les systèmes de gestion de la qualité
- Défaut de mise en œuvre des exigences applicables aux systèmes de gestion de la qualité
- Absence de mise en œuvre de mesures correctives et préventives appropriées lorsqu’une enquête sur les données post-commercialisation indique un schéma de défauts du produit
- Produits mis sur le marché entraînent un risque excessif pour le patient et/ou les utilisateurs lorsque le dispositif est utilisé conformément à l’étiquetage du produit.
- Existence de produits qui ne sont pas conformes aux spécifications du client et/ou aux exigences réglementaires
- Non-conformités répétées lors d’audits précédents
Cas où un audit inopiné (ou à court terme) est nécessaire
- Cas associés au SMQ :
- déficience significative constatée
- nouveau propriétaire
- extension du contrôle de la fabrication et/ou de la conception
- nouvelle installation, changement de site
- nouveaux processus, changements de processus
- modifications de l’autorité définie du représentant de la direction (le responsable qualité / affaires réglementaires)
- Cas associés au produit :
- nouveaux produits
- nouvelle catégorie de dispositifs
- Cas généraux :
- changements dans les normes, les règlements
- justifié par la surveillance postmarché, la vigilance
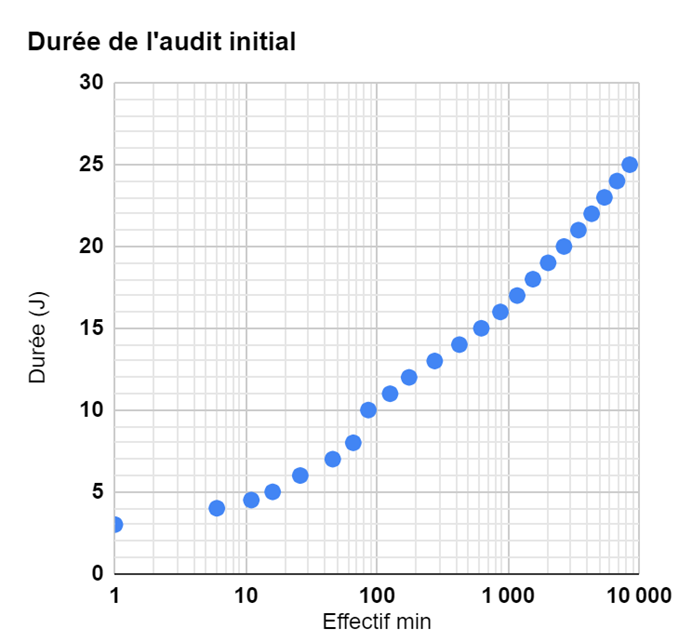
Durée de l’audit initial
Durée de base
Déterminée uniquement selon le nombre d’employés :
| Effectif (min) | Effectif (max) | Durée (jours) |
|---|---|---|
| 1 | 5 | 3 |
| 6 | 10 | 4 |
| 11 | 15 | 4,5 |
| 16 | 25 | 5 |
| 26 | 45 | 6 |
| 46 | 65 | 7 |
| 66 | 85 | 8 |
| 86 | 125 | 10 |
| 126 | 175 | 11 |
| 176 | 275 | 12 |
| 276 | 425 | 13 |
| 426 | 625 | 14 |
| 626 | 875 | 15 |
| 876 | 1175 | 16 |
| 1176 | 1550 | 17 |
| 1551 | 2025 | 18 |
| 2026 | 2675 | 19 |
| 2676 | 3450 | 20 |
| 3451 | 4350 | 21 |
| 4351 | 5450 | 22 |
| 5451 | 6800 | 23 |
| 6801 | 8500 | 24 |
| 8501 | 10700 | 25 |
Curieusement, cela ressemble à une loi logarithmique:
Au-delà de 10’700 employés, l’IAF invite à suivre la progression exposée :
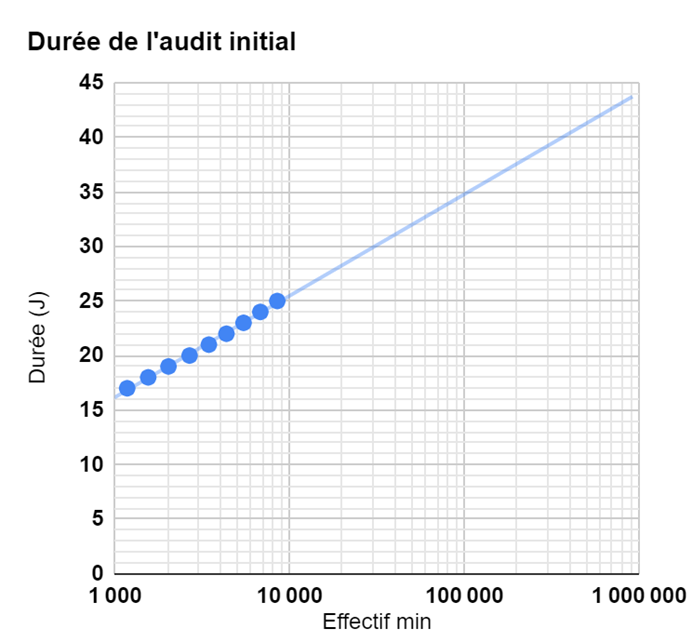
Soit un peu moins de 44 jours d’audit pour 1’000’000 de salariés.
En résumé: l’IAF invite à passer de 1109 ms à 24 heures, par membre de l’entreprise, selon la taille de la société.
Augmenter / Diminuer la durée de base
- Facteurs pouvant augmenter la durée d’audit : domaine technique, complexité du dispositif, complexité de la production, multiples activités, double certification ISO 9001 et ISO 13485
- Facteurs pouvant réduire la durée d’audit : pas de fabrication, pas de conception
Source : IAF