Avis des groupes d’experts sur les évaluations cliniques : procédure et synthèse des opinions
[article initialement publié le 6 juillet 2021, mises à jour régulières]
La “Procédure de consultation dans le cadre de l’évaluation clinique pour certains DM” est une exigence supplémentaire pour les évaluations cliniques des DM III implantables et IIb délivrant/retirant un médicament, selon l’article 54 du règlement (UE) 2017/745.
Cet article explique la procédure de consultation, détail le contenu d’un avis et résume les avis publiés. Il est mis à jour à l’occasion des nouvelles publications.
Procédure de consultation des groupes d’experts
Ci-dessous un rappel du process réglementaires, répété ad-libitum :
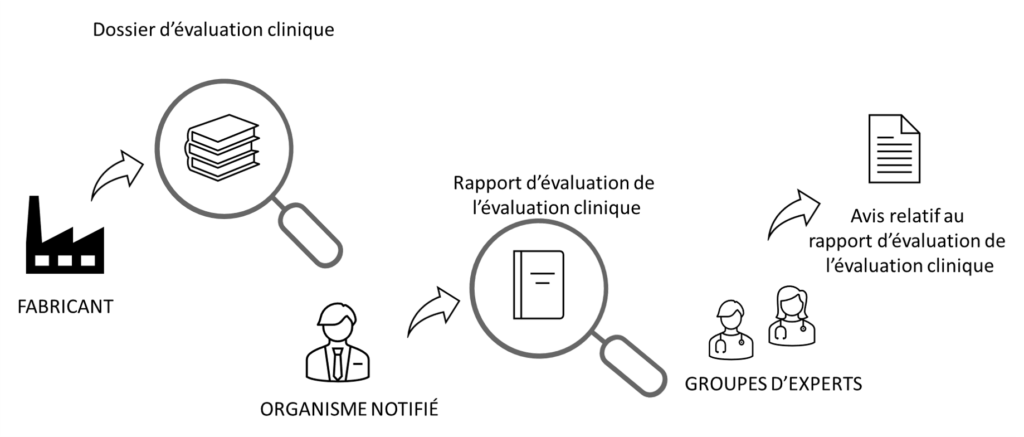
- Le fabricant produit une évaluation clinique
- L’organisme notifié du fabricant produit un rapport d’évaluation de l’évaluation clinique (selon le MDCG 2020-13)
- Le groupe d’experts européen :
- Évalue la nécessité de produire une opinion (étape “sélection”, par le groupe d’experts “Examen préliminaire“)
- Le cas échéant : produit une opinion sur le rapport de l’ON (étape “avis”, par le groupe d’experts le plus adapté au DM évalué)
- L’organisme notifié tient compte de l’opinion du groupe d’expert
- Le fabricant tient compte du fait que l’ON tienne compte de l’opinion du groupe d’expert
Une aide pour vos dossiers
Il peut être important, voire critique, de tenir compte des avis publiés, mêmes s’ils ne concernent pas directement vos produits. C’est d’autant plus nécessaire :
- Si vous êtes soumis à la procédure de consultation du groupe d’experts, pour comprendre leur attentes et angles d’approche
- Si vous êtes équivalent au dispositif visé : pour profiter de l’étude de l’état de l’art inclue au rapport (publications et problèmes sur le terrains)
- Si vous n’avez pas trop trop confiance en votre ON
👉 De manière générale, ces avis permettent de mieux comprendre les finalités des études cliniques.
Contenu d’un avis de groupe d’experts
Les § suivants reprennent le plan d’un avis

Partie 1 : Décision du groupe d’experts
Ici, la nécessité de produire une opinion est évaluée par le groupe “Examen préliminaire” selon 3 critères :
- Caractère nouveau et risques associés;
- Préoccupations sanitaires identifiées dans la littérature;
- Problème de vigilance sur le terrain (augmentation constatée des risques).
Résumé de la sélection
- Date de décision
- Le cas échéant : résumé des raisons si les informations sont jugées insuffisantes pour parvenir à une décision.
- Résumé des raisons de produire une opinion. Ceci peut être motivé par des incertitudes relatives :
- à l’utilisation prévue
- aux performances du dispositif
- à la qualité des données cliniques, notamment :
- durée des études
- exhaustivité, relativement aux indications d’utilisation
- Autres commentaires : il s’agit de développer les raisons qui ont confirmé/infirmé le besoin d’avis.
Les trois critères de sélection
Critère 1 : Nouveauté et impact
La nouveauté peut concerner les dispositif et/ou la procédure d’utilisation associée.
- Description de la nouveauté :
- indications d’utilisations
- procédures d’utilisations
- principe de fonctionnement
- composition physique
- process de fabrication
- ..
- Degré de nouveauté :
- ⭐⭐⭐Élevé
- ⭐⭐Moyen
- ⭐Bas
- Incertitudes liées à la nouveauté, ex : validité clinique sur le long terme
- Possibles impacts négatifs sur la santé associés à la nouveauté
- Gravité des impacts négatifs, sur 4 niveaux dont un nul :
- ⭐⭐⭐Majeur
- ⭐⭐Modéré
- ⭐Mineur
- Aucun
- Incertitudes liées à l’impact clinique, ex : incertitudes sur la durée, sur le protocole clinique, sur les indications …
Critère 2 : Préoccupations sanitaires
Ou, en version longue, “Préoccupations sanitaires scientifiquement valables conduisant à des modifications significativement défavorables du profil bénéfice/risque”.
- Biblio : la liste des publications utilisées : les sources des données
- Groupe / catégorie de DM : vraisemblablement selon la nomenclature EMDN
- Source de préoccupations sanitaires : pouvant porter sur
- les composants;
- les matériaux;
- l’impact sur la santé en cas de défaillance de l’appareil.
- Description des préoccupations et de l’impact sur la santé
- Qualité et pertinence des données utilisées
- Résumé des préoccupations sanitaires
Critère 3 : Augmentation significative des incidents graves
- Pertinence des données vis-à-vis du DM évalué
- Résumé des informations utilisées
Sélection du groupe d’experts
Le cas échéant, un panel d’experts est choisi pour formuler une opinion, parmi les dix groupes existants (cardio, endocrino, gastro…).
Partie 2 : Opinion du groupe d’experts
Résumé
👉 L’opinion statut sur l’adéquation de l’étude, attention : l’avis est très tranché, il sera vite défavorable s’il y-a trop de risques ou d’incertitudes dans le dossier.
- Description du DM : identification et indications médicales
- Caractère nouveau du DM : identification et degré de nouveauté
- Évaluation de l’évaluation : type d’évaluation clinique (nouvelle données, littérature); adéquation de l’étude (durée, indication, pertinence ds équivalence…); adéquation du SCAC planifié
- Adéquation des preuves cliniques : identification des manques de données cliniques, relatives aux risques comme aux bénéfices, en fonction des utilisations prévues
- Estimation du rapport B/R : juge si les conclusions sur le B/R sont recevables, au regard des éléments ci-dessus
- Preuves cliniques vs indications médicales : statut si les preuves sont suffisantes, en fonction des indications
- Planification du SCAC (PMCF) : évalue si le plan de SCAC est adapté
- Conclusions et recommandations : le cas échéant, liste des conseils pour reprendre l’évaluation clinique, formulés au conditionnel mais à prendre à l’impératif
Détails
C’est ici que l’évaluation de l’ON est réellement critiquée, selon cinq critères :
- Avis sur l’évaluation par l’ON du caractère adéquat du rapport d’évaluation clinique du fabricant.
- Avis sur l’évaluation par l’ON du caractère suffisant des preuves cliniques fournies par le fabricant.
- Avis sur l’évaluation par l’ON du caractère adéquat de la détermination du rapport bénéfice/risque par le fabricant.
- Avis sur l’évaluation de l’ON de la cohérence des preuves cliniques du fabricant avec l’objectif visé, y compris la ou les indications médicales
- Avis sur l’évaluation par l’ON de la cohérence des preuves cliniques du fabricant avec le plan de SCAC.
👉 Notez que la liste des publications utilisées pour forger l’avis est fournie.
Conclusions générales et recommandations
Une synthèse de l’avis du groupe d’expert, vous pouvez commencer par ce résumé pour vous faire une idée de l’avis.
Divergences dans le groupe d’experts
Le cas échéant, les divergences entre les experts sont expliquées.
Liste des avis publiés
Le tableau ci-dessous fait une synthèse des opinions publiées, un lien est fourni vers chacun des rapports.
| 06/15/2021 | 08/19/2021 | 12/07/2021 | 05/23/2022 | 06/27/2022 | 10/06/2022 | 08/01/2022 | 09/23/2022 | 10/28/2022 | 11/11/2022 | 06/10/2024 | 06/15/2024 | 11/18/2024 | 12/05/2023 | 04/07/2025 | |
| Link | https://health.ec.europa.eu/system/files/2021-12/cepc-2021-000201_opinion_en.pdf | https://health.ec.europa.eu/system/files/2021-12/cecp-2021-000205_opinion_en.pdf | https://health.ec.europa.eu/system/files/2021-12/cecp-2021-000207_opinion_en.pdf | https://health.ec.europa.eu/system/files/2022-07/cecp-2022-000213_opinion_en.pdf | https://health.ec.europa.eu/system/files/2022-08/cecp-2022-000216_opinion_en.pdf | https://health.ec.europa.eu/system/files/2022-10/cecp-2022-000227_opinion_en_0.pdf | https://health.ec.europa.eu/system/files/2022-10/cecp-2022-000222_opinion_en.pdf | https://health.ec.europa.eu/system/files/2023-03/cecp-2022-000225_opinion_en.pdf | https://health.ec.europa.eu/system/files/2022-12/cecp-2022-000232_opinion_en.pdf | https://health.ec.europa.eu/document/download/91498a0c-1f12-4dc0-a6fc-9c4d40f0ccbd_en | https://health.ec.europa.eu/document/download/28b1bf28-3153-43ea-92fb-58799a09dd61_en | https://health.ec.europa.eu/document/download/a274863e-c578-4f6d-a525-1c3b5c0aebb6_en | https://health.ec.europa.eu/document/download/80f2432d-a9da-48ae-8ebb-0054b4922033_en | https://health.ec.europa.eu/document/download/eaaa5294-7fde-4c64-8c91-25c023035570_en | https://health.ec.europa.eu/document/download/0ad86a3e-8bcd-4d3b-b24b-588f74ab294e_en |
| MD type | bone graft material | acetabular inserts/cup | artificial plumonary heart valve | transcatheter heart valves | tricuspid valve replacement system | Resorbable mesh with a resorbable hydrogel coating | Implantable stimulation device for neurological applications | Iron alloy bioabsorbable stent. | Anatomic shoulder prosthesis | Single chamber, extravascular implantable cardioverter defibrillator (ICD) |
Bioabsorbable mesh | Drug eluting stent | Mechanical Ventilator | Male genital system prostheses (MALE GENITAL SYSTEM PROSTHESES) – hyaluronic acid (sodium salt) gel for intralesional penile injection | Structures filling, replacement and reconstruction devices — Intended purpose: Intraarticular joints viscosupplementation |
| Class | III | III | III | III | III | III | III | III | III | III | III | III | III | IIb | III |
| Expert planel | General and plastic surgery dentistry | Orthopaedics, traumatology, rehabilitation, rheumatology |
Circulatory system | Circulatory system | Circulatory system | General and plastic surgery and dentistry | Neurology | Circulatory system | Orthopaedics, traumatology, rehabilitation, rheumatology | Circulatory system | General and plastic surgery and dentistry | Circulatory system | Respiratory, anaesthesiology, intensive care |
Nephrology and urology | Orthopaedics, traumatology, rehabilitation, rheumatology |
| Competence area | Maxillofacial surgery & Dentistry | Joint replacements (hip, knee, shoulder) | Prosthetic heart valves and devices for heart valve repair | Prosthetic heart valves and devices for heart valve repair | Prosthetic heart valves and devices for heart valve repair | Surgical implants and general surgery | Central and peripheral nervous system devices | Prosthetic heart valves and devices for heart valve repair | Non-loadbearing joint replacements | Active implantable cardiac devices and electrophysiological devices | Surgical implants and general surgery | Cardiovascular stents (metallic and bio-resorbable) and vascular prostheses | Respiratory and anaesthetic devices | Devices for nephrology and urology | Orthopaedics, traumatology, rehabilitation, rheumatology |
| Implantable ? (yes/no) | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | No | yes |
| NB name | MDC MEDICAL DEVICE CERTIFICATION GMBH | BSI Group The Netherlands B.V. | DEKRA Certification B.V. | DEKRA Certification B.V. | DEKRA Certification B.V. | BSI Group The Netherlands B.V. | DEKRA Certification B.V. | DEKRA Certification B.V. | GMED SAS | TÜV SÜD | BSI Group The Netherlands B.V. | 3EC | UDEM Adriatic d.o.o., | Eurofins Product Testing Italy S.r.l. | POLSKIE CENTRUM BADAN I CERTYFIKACJI S.A. |
| Novelty Type (Device, Procedure, Device and Procedue) | Device | Device | Device | Procedure | Device and Procedure | None | Device and Procedure | Device | None | Device and Procedure | None | Device | none | procedure | Device |
| Novelty Level (none, low, medium, high) | Medium | Low | High | High | High | None | High | High | None | High | None | Medium | none | high | high |
| Clinical impact (none, minor, moderate, major) | Moderate | Minor | Major | Major | Major | None | Major | Major | None | Moderate clinical or health impact | No clinical or health impact | Major clinical or health impact | Major clinical or health impact | Major | moderate |
| Overall conclusions and recommendations | Presented clinical data for one indication (#4) are presently insufficient and should be extended to include at least the healing phase for the implant (additional 4 months) and the results can then also be used for a positive clinical assessment for indication #3 and #5. For the other indications data from clinical studies are missing and therefore the evidence for these indications in insufficient. Literature survey is flawed by the fact that the new device is similar but not equivalent to market products. The PMCF plan needs to be extended and specified e.g. to cover in detail the other claimed indications. If relevant data are available, the indications can be accordingly extended. | The expert panel concurs with the NB’s assessment of the manufacturer’s conclusions on benefit-risk determination for the change in PMMA spacer material grade. Longitudinal monitoring of clinical outcomes as proposed in the manufacturer’s PMCF plan is considered adequate to detect unexpected mechanisms of failure, insufficient performance, and a survival rate below the acceptable rate for clinical excellence. | The manufacturer should review the clinical and market availability of similar devices for the same indications as expressed in the Valve System IFU. Specifically, the expert panel identified one medical device approved by the U.S. Food and Drug Administration for clinical use. The CER should discuss differences and similarities of the devices and include another device clinical evidence into the CER discussion. Misinformation on Valve System generations should be clarified, with specific indications on which generation types have been used in studies presented in CER, the reasons for generation improvements and the existence of the 4th device generation for clinical use in a clinical research study published in 2021 |
Patients at high risks : The panels agrees with the NB.Patients at low to intermediate risks : The panel considers that the benefit/risk balance of alves for this subpopulation is not sufficiently supported by evidence in terms of amount and quality. The panel recommends that more prolonged follow-up from AVIV is reported (especially as the database extract was last performed in August 2020) and that additional studies are conducted so as to confirm external validity of the first registry. | A positive benefit-risk ratio is not sufficiently demonstrated. A randomized controlled trial with comparison to optimized medical therapy alone would be relevant to properly assess the efficacy and safety of this novel technology. | Expert panel agrees with the NB’s assessment on the benefit-risk ratio of this device for the clinical indications currently under scrutiny in the early postoperative period. Further long-term studies with appropriate follow-up strategies are needed to draw definitive conclusions. | Details with similar accuracy should be provided on AE. Details on the stimulation procedures should be considered as for direct stimulation (…) the electrode positioning might be crucial for the success of the approach and thus for the risk/benefit analysis. It is not clear whether a system or procedure is in place to define the stimulation setting. No post-market surveillance data is available at this stage. The NB may have overlooked the fact that the medical indications need to be further developed (…) | 2 major concerns: 1. Indication: the intended purpose should be restricted, 2. Uncertainty about the clinical benefit: There is no data about the benefit compared over other therapeutic options. Advantage over other devices cannot be estimated | Data of the shoulder implant system under review does not meet minimum standards when compared to state-of-the-art implants of the similar group of shoulder implants in particular revision rates and complications (…) A well-designed PMCF clinical protocol was not executed as planned, Instead, an internal retrospective dataset of shoulder implant patients was presented, with limited follow-up, limited patient number and considerable numbers of loss-to-follow-up. The planned 10-year clinical dataset for September 2022 is not provided in the CER. Finally, the literature search was not adequate as it only retrieved 3 articles, while at least 5 additional articles were found at the time of the literature search performed by the manufacturer. | Data provided by the manufacturer for the initial submission was adequately assessed by the NB and confirmed that the device can be considered as an option for patients with indication for ICD therapy, in particular S-ICD, who could also benefit from ATP. It is recommended that manufacturers and NBs update their clinical reports with the latest relevant clinical information as it becomes available, in particular the incidence of major complications related to the device. In this case, the lack of up-to-date clinical data might have led to partial conclusions and possibly to an underestimation of the true clinical risks (e.g., rates of inappropriate shocks, infections, lead dislodgment). In addition, the rate of 25% for ATP switching off, in many cases due to intolerability (at least 15% of the patients did not accept the pacing threshold testing), the limited efficacy of contactless ATP and the potential contraindications to future epicardial access for VT ablation need to be considered for the clinical indications. Possible limitations in patients with known and frequent sVT also needs to be considered. The acquisition of more long-term safety data is highly recommended. | The NB has performed an in-depth analysis of the documents provided by the manufacturer and has documented their conclusion in the CEAR. The manufacturer demonstrated benefit of the prophylactic use of meshes by providing a thorough and detailed literature review. The literature review takes into consideration three publications that address the specific usage of the device under assessment. Additionally, the manufacturer provided real-world evidence and data from one investigator-sponsored trial. A few shortcomings have been identified in relation to the newly added indication. The data provided has a limited follow-up period (for most patients, only 30 days, and only for a limited number of patients one year or longer). The following limitations have been observed: 1. There is a limited number of patients (51) that have data beyond 30 day follow-up, although only limited adverse events and complications have been reported. Further research in the long-term usage (up to 5 years) is necessary to confirm the conclusion on the benefit-risk profile of the device under assessment with the new indication of prophylactic use. 2. No data has been provided in which the device under assessment is compared to a nonresorbable device that is established for a prophylactic indication. | The expert panel opinion is in agreement with the assessment performed by the NB but recommends a longer follow up period of the patients enrolled in the DESyne BDS Plus RCT study than currently foreseen in the PMCF plan, in order to investigate long-term risks of using rivaroxaban and argatroban as ancillary medicines in this device. |
This expert panel found the following serious issues with both the documents presented by the NB for the clinical assessment and the documents presented by the manufacturer to the NB: – Inconsistencies were identified in those documents, namely between the Clinical Evaluation Plan {CEP), Clinical Evaluation Report (CER) from the manufacturer and the Clinical Evaluation Assessment Report, {CEAR), affecting, among other things, properties and parameters critical for assessing clinical adequacy and safety. – The clinical evaluation is supported by a literature review that is limited both in scope and applicability to the specific device under consideration. Additionally, equivalence is claimed to an already marketed device. However, the clinical equivalence cannot be properly reviewed because of the inconsistencies mentioned (it is not clear to which device is the equivalence claimed), and incomplete documentation provided. Therefore, the panel considered there was insufficient information to allow a thorough assessment of the clinical impact of the device. – There were very few details provided in the Post-Marketing Clinical Follow-Up plan (PMCF plan). Given the uncertainties highlighted concerning the data provided for the pre-market clinical evaluation, the lack of details on the PMCFplan becomes even more significant to be able to evaluate the need for additional data in the post-marketing phase. The experts are particularly concerned about several issues related to the clinical evaluation, namely: Insufficiency of the evidence about the safety of the device; Absence of evidence that a meaningful clinical evaluation was performed and the lack of evidence supporting the adequacy of the device for the intended purpose and the chosen target population; Serious mistakes on the pathophysiology of mechanical ventilation and its benefits but also on the risks for the patients; Absence of a comprehensive PMCFplan; The failure of the NB to detect and adequately address the issues explained in the previous points .Moreover, the documents contained numerous errors and inconsistencies and overall were difficult to understand. (…)Therefore, the expert panel has concerns that the NB assessment of the CER has been adequately performed and that it has demonstrated the benefit risk profile of the device. | The CER and the NB’s CEAR thoroughly cover the literature and the rationale/posology, but the panel identifies major gaps regarding evidence robustness, especially for mid-/long-term outcomes. Key issues: short follow-up (often 3 months), lack of sham control, absence of dose–response data, lack of data on the fate of the product (degradation profile, migration), and insufficient discussion/control of the risk of inadvertent intracavernosal/intravascular injection, although the IFU includes a warning. Evidence supports use exclusively in the acute phase (no evidence for chronic phase). The PMCF plan is broadly consistent, but longer follow-up is desirable. Recommendations: provide additional information on potential pharmacological effects of HA; document the degradation profile and migration; strengthen instructions and warnings to avoid intravascular injection and to guide proper intralesional injection; complement PMCF with a multicentre registry with long-term follow-up, and extend follow-up of the post-market study (12–24 months desirable). The panel raises concerns and requests additional information, notably on long-term benefit–risk and management of intravascular injection risk; no explicit “validation” of NB conclusions | The panel considers that the clinical evidence presented does not adequately establish safety and clinical performance, particularly because the study was a prospective, open-label, non-randomized clinical trial without a control group and using subjective criteria that were subject to significant placebo bias. It also considers that a 6-month follow-up period is insufficient given the novelty of the treatment. Key recommendation: require a randomized controlled trial (RCT) with a follow-up period of at least 1 to 2 years to robustly demonstrate safety and clinical performance. |
| Conclusions of the notified body validated ? (yes/no) | no | yes | no | no | no | yes | no | no | no | yes | yes | yes | no | no | no |
Source : Groupes d’experts MD UE