Focus : Période de transition
- Contexte
- Échéances
- Obligations
Vers le règlement (UE) 2017/745
Le règlement (UE) 2017/745 relatif aux dispositifs médicaux, remplaçant les directives 90/385/CEE et 93/42/CE, répond à 3 besoins :
- Simplifier la règlementation
- Durcir les exigences sur les produits et les processus
- Éviter un nouveau scandale PIP
Le scandale PIP a 3 causes :
- Les pratiques criminelles d’un fabricant
- L’incapacité de l’organisme notifié à détecter les fraudes
- L’inefficacité des autorités compétentes
Le règlement (UE) 2017/745 a connu 3 périodes de transitions :
- Une première prévue par le règlement de 2017
- Une seconde en réponse à la crise du coronavirus
- Une troisième en réponse aux risques de pénuries de dispositifs médicaux en Europe
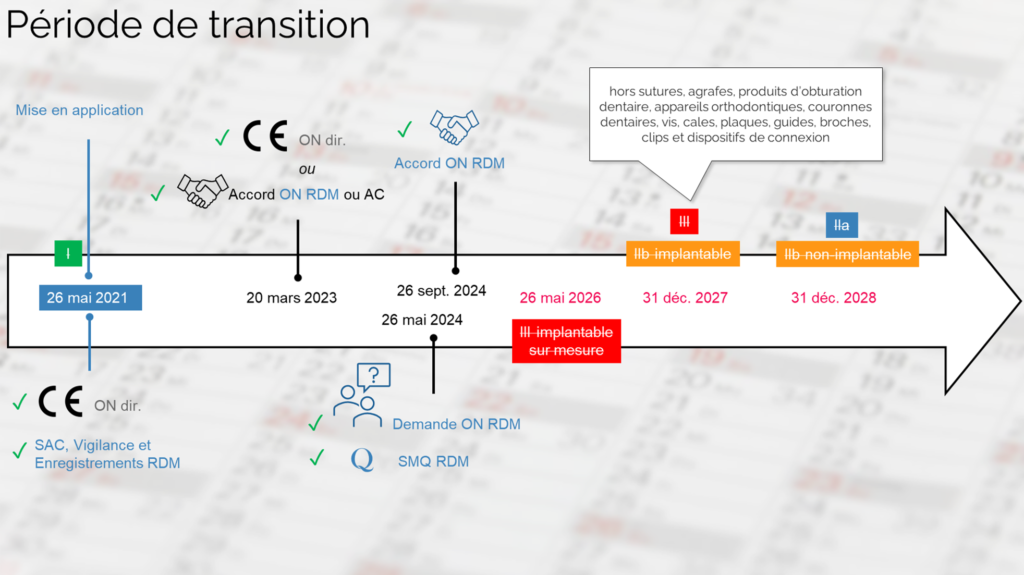
Les dispositions transitoires pour les dispositifs médicaux déjà CE sous une directive s’étendent jusqu’en 2029 :
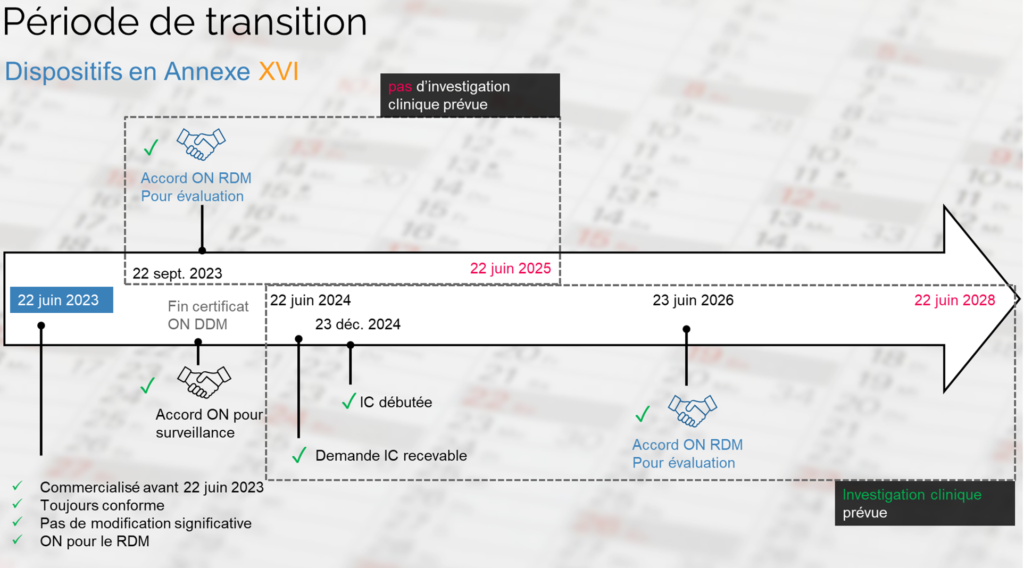
Les dispositions transitoires pour les dispositifs en annexe XVI varient selon que vous ayez ou non planifié une investigation clinique :
Pour profiter du prolongement d’un marquage CE directive, les fabricants de DM doivent respecter des conditions :
- Les dispositifs ne doivent pas présenter de risque inacceptable
- Les dispositifs doivent rester conforme à la directive
- Pas de changement significatif dans la conception et la finalité
- Au + tard le 26 mai 2024 : mise en place d’un SMQ selon l’article 10.9
- Au + tard le 26 mai 2024 : demande formelle pour l’évaluation de la conformité par un ON
- Au + tard le 26 sept 2024 : accord écrit signé avec l’ON.
- Le fabricant doit respecter les exigences du RDM en matière de SAC, de vigilance et d’enregistrement
L’article 10.9 du règlement détaille les exigences sur le SMQ :
Le système de gestion de la qualité porte au moins sur les aspects suivants :
a) une stratégie de respect de la réglementation, notamment le respect des procédures d’évaluation de la conformité et des procédures de gestion des modifications apportées aux dispositifs concernés par le système ;
b) l’identification des exigences générales en matière de sécurité et de performances et la recherche de solutions pour les respecter;
c) la responsabilité de la gestion;
d) la gestion des ressources, et notamment la sélection et le contrôle des fournisseurs et sous-traitants;
e) la gestion des risques visée à l’annexe I, section 3;
f) l’évaluation clinique conformément à l’article 61 et à l’annexe XIV, y compris le SCAC;
g) la réalisation du produit, y compris la planification, la conception, l’élaboration, la production et la fourniture de services;
h) la vérification des attributions d’IUD conformément à l’article 27, paragraphe 3, à l’ensemble des dispositifs concernés en veillant à la cohérence et à la validité des informations fournies conformément à l’article 29;
i) l’élaboration, la mise en œuvre et le maintien d’un système de surveillance après commercialisation conformément à l’article 83;
j) la gestion de la communication avec les autorités compétentes, les organismes notifiés, les autres opérateurs économiques, les clients et/ou d’autres parties prenantes;
k) les processus de notification des incidents graves et des mesures correctives de sécurité dans le cadre des activités de vigilance;
l) la gestion des mesures correctives et préventives et la vérification de leur efficacité;
m) les processus de surveillance et de mesure des résultats, d’analyse des données et d’amélioration des produits.