Les rôles et obligations dans le Règlement (UE) 2017/745
Le nouveau règlement (UE) 2017/745 clarifie les obligations des différents acteurs de la vie d’un dispositif médical.
Cet article reprend les définitions et exigences applicables aux fabricants, mandataires, importateurs et distributeurs.
4 rôles possibles
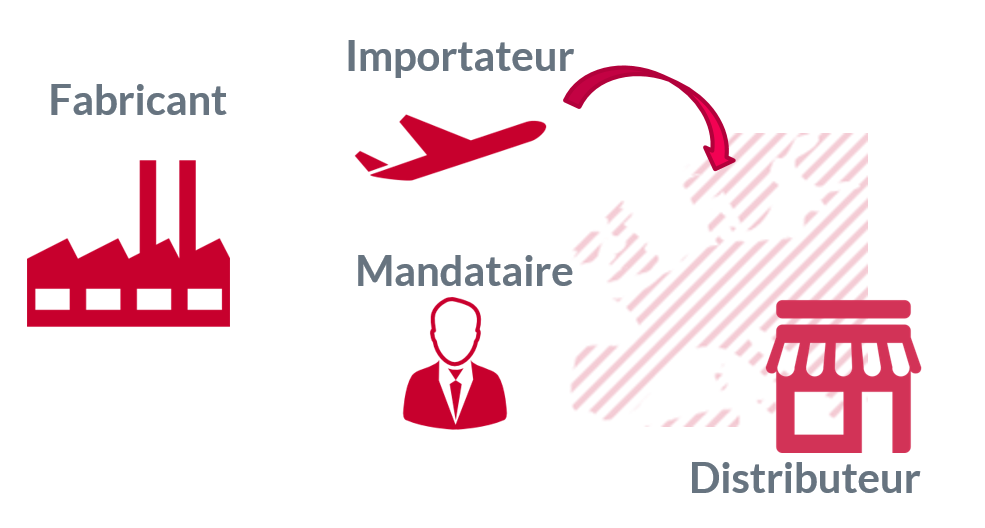
Le règlementent identifie jusqu’à 4 rôles, tous ne sont pas nécessaires en fonction des cas et un même acteur peut jouer différents rôles.
Dans le cas le plus simple : un fabricant Français n’a pas besoin d’importateur ni de mandataire et peut très bien assurer la distribution du dispositif.
Dans le cas le plus complexe :
- Un fabricant hors EU est responsable du marquage CE de son produit.
- Un importateur met le dispositif sur le marché de l’UE.
- Un mandataire agit au nom du fabricant pour les taches règlementaires européennes.
- Un distributeur met le dispositif à disposition de l’utilisateur.
Le règlement insiste sur une bonne coopération pour la surveillance des dispositifs, sur une nécessité de se “surveiller les uns les autres” et de remonter les informations de surveillance post-commercialisation au fabricant et (lorsque nécessaire) aux autorités nationales.
1. Le Fabricant

Définition
Fabricant : toute personne physique ou morale qui fabrique ou remet à neuf un dispositif ou fait concevoir, fabriquer ou remettre à neuf un dispositif, et commercialise ce dispositif sous son nom ou sous sa marque
Notez que le fabricant ne fabrique pas forcément le dispositif : fabrication et conception peuvent être sous-traités (cas de l’OBL / OEM), en d’autre mot : “c’est celui qui est sur l’étiquette qui est responsable”.
Obligations
L’essentiel du règlement vise le fabricant, ses obligations sont définies aux chapitres II (mise à disposition des DM), III (identification et traçabilité) et V (évaluation de la conformité). Les annexes viennent en complément.
2. Le Mandataire

Définition
Mandataire : toute personne physique ou morale établie dans l’Union ayant reçu et accepté un mandat écrit d’un fabricant, situé hors de l’Union européenne, pour agir pour son compte aux fins de l’accomplissement de tâches déterminées liées aux obligations incombant à ce dernier en vertu du présent règlement
Les mandataires sont utiles aux fabricants hors UE, c’est le mandataire qui est tenu, par contrat, d’agir à sa place vis-à-vis des obligations réglementaires (ex : effectuer les déclarations de mise sur le marché).
La désignation d’un mandataire est obligatoire, son acceptation par écrit constitue également une exigence.
Obligations
Elles sont définies dans l’article 11 :
- Vérifier le marquage CE : le fabricant doit avoir établi une déclaration de conformité UE, avoir constitué la documentation technique du DM, avoir appliqué la bonne procédure de marquage CE.
- Tenir à la documentation à disposition des autorités nationales : le dossier technique, la déclaration de conformité et, le cas échéant, le certificat de conformité délivré par un organisme notifié.
- Obtenir un numéro unique d’identification (UDI) du dispositif.
- Faire le lien fabricant <=> autorité compétente (demande d’échantillons, gestion des risques, signalement d’incidents).
- Mettre fin au mandat si le fabricant ne respecte pas ses obligations et tenir informées les autorités compétentes.
3. L’Importateur

Définition
Importateur : toute personne physique ou morale établie dans l’Union qui met un dispositif provenant d’un pays tiers sur le marché de l’Union”
L’importateur est le plus souvent mandataire et/ou distributeur.
Obligations
D’après l’article 13 :
- Vérifie la présence du marquage CE et de la déclaration de conformité, la bonne désignation d’un mandataire, la conformité de l’étiquetage, la présence d’une notice d’utilisation, l’attribution et l’enregistrement d’un IUD (Identifiant Unique du Dispositif) auquel il associe ses coordonnées.
- En cas de problème de conformité du dispositif : l’importateur informe le fabricant (et son mandataire) avant de refuser l’importation ; en cas de risque grave ou de falsification, il doit également en informer l’autorité compétente.
- Indique ses coordonnées sur l’emballage du DM ou sur la documentation.
- Vérifie les conditions de transport et de stockage.
- Tient un “registre de plaines” : produits non conformes, rappels et retraits.
- Collabore avec le fabricant (le mandataire) et les autorités compétentes en cas d’investigation des plaintes.
- Informe le fabricant en cas de non-conformité, d’incidents.
4. Le Distributeur

Définition
Distributeur : toute personne physique ou morale faisant partie de la chaîne d’approvisionnement, autre que le fabricant ou l’importateur, qui met un dispositif à disposition sur le marché, jusqu’au stade de sa mise en service”
Les boutiques de matériel médical, pharmacies, sites de vente en ligne, supermarché, magasins de sport, … le panel est très large, voire surprenant.
Obligations
Voir l’article détaillé sur les obligations des distributeurs.
Elles sont définies dans l’article 14 :
- Vérifier que le dispositif porte le marquage CE, que la déclaration CE a été établie, que les documents à l’intention de l’utilisateur sont disponibles, que l’IUD a été attribué, le cas échéant : que l’importateur est conforme aux exigences du règlement.
- Informer le fabricant / importateur/ mandataire en cas de non-conformité.
- Informer le fabricant / importateur/ mandataire / les autorités compétentes en cas de risque grave ou de falsification.
- Veiller au respect des conditions de stockage et de transport des dispositifs.
- Effectuer la surveillance post-market : transmettre les signalements au fabricant / importateur / mandataire, gérer un registre de plaintes / non-conformités / rappels / retraits.
- Le cas échéant, communiquer aux autorités réglementaires les documents demandés.
26 commentaires