L’ISO 14971 à l’heure du règlement DM 2017/745

Le Règlement (UE) 2017/745 relatif aux dispositifs médicaux va quelque peu changer les habitudes en matière de gestion des risques.
Si l’application de la norme ISO 14971 est toujours d’actualité, les fabricants devront mettre à jour leurs procédures et documentations pour tenir compte du règlement. Celui-ci :
- introduit des exigences supplémentaires par rapport à la 14971.
- a quelques “écarts de contenu” avec la norme qu’il faudra respecter, les exigences règlementaires prenant le pas sur les exigences normatives.
Cet article met en regard les chapitres de la norme et les exigences du règlement.
Un tableau liste les écarts, il n’a rien d’officiel, il faudra attendre l’annexe Z de la future EN ISO 14971 pour être fixé.
1. Domaine d’application
Le règlement et la norme convergent : il s’agit d’identifier et de maitriser les risques associés aux dispositifs médicaux pendant tout le cycle de vie du dispositif.
2. Termes et définitions
Les bases sont communes avec quelques définitions propres au règlement, notamment celle de “détermination du rapport bénéfice/risque“.
La future révision de la 14971 devrait mieux tenir compte des bénéfices et introduire une définition tournant autour d’un impact positif / désirable; sur la santé du patient / sa prise en charge.
3. Exigences relatives à la gestion des risques
Exigence commune : mettre en œuvre un système continu et itératif de gestion des risques.
La direction devra décider d’une politique d’acceptabilité des risques en accord avec la règlementation applicable.
Ci-dessous un exemple de politique d’acceptation des risques, qui se veut en accord avec le règlement (mais encore une fois : à confirmer).
Un risque sera accepté à titre individuel si :
- il est maitrisé (annexe I.I.8 ” Tous les risques connus et prévisibles sont réduits au minimum“)
- son rapport B/R est favorable (annexe I.I.1 : “(…) risques acceptables au regard des bénéfices pour le patient“)
- il n’est plus possible de poursuivre la maitrise du risque sans affecter le rapport B/R global (annexe I.I.2 : “réduire les risques autant que possible signifie réduire les risques autant que possible sans altérer le rapport bénéfice/risque“)
L’ensemble des risques sera accepté si :
- le rapport B/R de chacun des risques est favorable; et
- le rapport bénéfices / ensemble des risques résiduels est favorable
(annexe I.I.4 : “de sorte que le risque résiduel associé à chaque danger ainsi que le risque résiduel global soient jugés acceptables“).
4. Analyse des risques
Le règlement demande de prendre en compte les “risques associés à l’utilisation prévue et à une mauvaise utilisation raisonnablement prévisible” (annexe I.I.3.c).
L’ IEC 62366-1 (ingénierie de l’aptitude à l’utilisation) permet de gérer les risques liés à une mauvaise utilisation.
L’EN ISO 14971:2012 évoque (timidement) la révision IEC 62366:2007, la prochaine révision de la 14971 appellera l’IEC 62366-1:2015 de façon plus explicite.
5. Évaluation des risques
Côté 14971 : c’est ici que le fabricant évalue si les risques sont à maitriser.
La probabilité et la gravité déterminées durant l’estimation sont utilisées pour évaluer le niveau de risque.
Exemple classique :
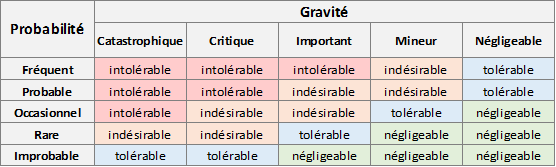
Une approche courante consiste à évaluer l’acceptabilité d’un risque en fonction de son niveau, les risques négligeables sont alors acceptés dès la première évaluation, sans mesure de réduction.
C’est beaucoup moins rigolo avec le règlement qui demande de réduire tous les risques au minimum.
Mais l’évaluation du niveau de risque n’est pas vaine pour autant, elle vous permet :
- De dimensionner les mesures de maitrise en fonction du niveau de risque à abaisser.
- De quantifier le risque pour l’étude du rapport B/R.
- D’accepter le risque (dans certains cas, voir plus bas).
- De planifier le suivi des tendances après commercialisation, en comparant niveau évalué et niveau réel.
- De sélectionner les risques qui méritent une surveillance accrue.
- D’avoir un indicateur pertinent pour votre processus de gestion des risques (ex : indicateur : niveau des risques résiduels, objectif : 90% ou plus sont négligeables)
- …
Maitriser un risque négligeable ?
Plusieurs options pour maitriser ce qui est pourtant négligeable :
- maitrise par information fournie à l’utilisateur (de toute façon le règlement le demande, mais les instructions vont rapidement devenir illisibles)
- maitrise par sensibilisation durant la formation de l’utilisateur
- maitrise par mise en œuvre du processus de gestion des risques : le risque a été identifié, évalué, il sera suivi. (un peu tiré par les cheveux)
- …
Et d’autres options pour justifier une non maitrise :
- prouver qu’aucun dommage n’est possible ou que la probabilité est nulle (dans ce cas, par définition, ce n’est plus un risque)
- démontrer l’impossibilité d’améliorer le rapport B/R (c’est rare)
- prouver que le niveau est déjà minimal et que l’ajout de mesures ne pourrait que dégrader les performances en complexifiant le dispositif
- préciser dans la politique qualité que les risques usuels / acceptés par la société ne sont pas à maitriser (ex : risques associés aux ondes électromagnétiques des dispositifs embarquant un modem radio). On se rabat ainsi sur l’état de l’art en matière d’acceptabilité des risques.
- …
6. Maîtrise des risques
Toujours l’écart “si une réduction s’impose” (norme) vs “tous les risques sont réduits” (règlement).
Concernant les options de maitrise : la maitrise via les activités de production est mise en première option dans le règlement, en seconde dans la norme.
Pour le choix des mesures de maitrise : le règlement demande de prendre en compte l’état de l’art (annexe I.I.4) ce qui sort du domaine d’application de la 14971 mais vous avez surement une procédure “conception et développement” qui vous le rappelle.
L’annexe I.I.5 développe des exigences pouvant être respectées en mettant en œuvre l’IEC 62366-1 pour gérer les erreurs d’utilisation : on s’appuiera sur la spécification d’utilisation établie durant l’IAU pour tenir compte du contexte; les caractéristiques ergonomiques sont-elles décrites dans les interfaces utilisateur relatives à la sécurité.
7 Évaluation de l’acceptabilité du risque résiduel global
Informations fournies à l’utilisateur
La 14971 laisse au fabricant le choix des informations à fournir pour maitriser les risques résiduels.
Le règlement demande d’indiquer tous les risques résiduels :
“Les fabricants informent les utilisateurs concernant tout risque résiduel” (annexe I.I.4)
“La notice d’utilisation contient toutes les informations suivantes : tout risque résiduel (…)” (annexe I.III.23.4.g)
Et les exigences règlementaires ne concernent pas que l’utilisateur/patient :
“(…) les méthodes d’élimination des risques auxquels sont exposées les personnes intervenant dans l’installation, l’étalonnage ou la maintenance du dispositif” (annexe I.III.23.4.g)
8 Rapport de gestion des risques
Pas d’exigence règlementaire explicite mais la mise en conformité demande nécessairement un dossier de gestion des risques, à inclure à la documentation technique du dispositif.
9 Informations de production et de postproduction
Ce dernier article est très timide dans la 1497, le règlement est plus exigent (la PMS est un des gros changements par rapport à la directive 93/42/CEE) :
- Mise à jour de la gestion des risques en fonction des informations de prod. et PMS (surveillance après commercialisation) :
- Annexe I.I.3;e : “(…) évaluent l’incidence des informations issues de la phase de prod. et de la PMS, sur les dangers et la fréquence, sur les estimations des risques, ainsi que sur le risque global, le rapport bénéfice/risque et le caractère acceptable du risque“
- Article 83.3.a : “actualiser la détermination du rapport bénéfice/risque et améliorer la gestion des risques“
- Suivi des tendances : annexe III.1.1.b : “Le plan de surveillance après commercialisation comprend au moins des indicateurs et des seuils adaptés à utiliser pour procéder à la réévaluation continue de l’analyse bénéfice/risque et de la gestion des risques“
Évaluation clinique
À peine évoquée dans l’ISO 14971, l’évaluation clinique utilise et nourrit pourtant le processus de gestion des risques :
- Les données cliniques apportent le niveau de preuve suffisant pour garantir les bénéfices et la maitrise des risques.
- L’investigation clinique utilise la gestion des risques pour garantir un rapport B/R favorable, durant l’investigation et après commercialisation.
Article 61.1 : “La confirmation de la conformité aux exigences générales pertinentes en matière de sécurité et de performances (…) ainsi que l’évaluation des effets secondaires indésirables et du caractère acceptable du rapport bénéfice/risque (…), sont fondées sur des données cliniques apportant une preuve clinique suffisante”
Article 62.1.c : “Les investigations cliniques sont conçues (…) en vue de l’évaluation de la conformité, aux fins de l’un des objectifs suivants (…) établir et vérifier la sécurité clinique du dispositif et détecter les éventuels effets secondaires indésirables dans des conditions normales d’utilisation du dispositif et évaluer si ceux-ci constituent un risque acceptable au regard des bénéfices attendus du dispositif concerné”
Tableau : Écarts règlement 2017/745 / ISO 14971:2007
| ISO 14971:2007 | règlement 2017/745 |
|---|---|
| Si une réduction du risque s’impose, les activités de maîtrise du risque doivent (…) (6.1) | Tous les risques connus et prévisibles ainsi que tous les effets secondaires indésirables sont réduits au minimum. (A I.I.8) |
| a) sécurité inhérente par conception; b) mesures de protection (…) ou au sein du processus de fabrication; (6.2) |
a) éliminer ou réduire les risques autant que possible grâce à une conception et une fabrication sûres; (A I.I.4) |
| Si le risque résiduel n’est pas jugé acceptable (…) et qu’il ne soit pas possible de poursuivre un cycle de maîtrise des risques, le fabricant peut (…) déterminer si les bénéfices sont supérieurs au risque. (6.4) | (…) étant entendu que les risques éventuels liés à leur utilisation constituent des risques acceptables au regard des bénéfices pour le patient. (A I.I.1) |
| Le fabricant doit décider (…) des informations qui doivent figurer dans les documents d’accompagnement, afin d’indiquer ces risques résiduels. (6.4) | La notice d’utilisation contient toutes les informations suivantes (…) tout risque résiduel (…) ainsi que les informations à transmettre au patient à cet égard. (A I.III.23.4.g) |
Notez que certains écarts de la 93/42 (expliqués dans l’annexe Z) semblent résolus (recours aux informations fournies pour maitriser les risques et obligation de mettre en œuvre toutes les options de maitrise).
Sur le même sujet : vous pouvez consulter l’article listant les documents et normes sur la gestion des risques des DM.
6 commentaires