ISO 14971 : Gestion des risques des dispositifs médicaux – Introduction

La norme ISO 14971 est une norme harmonisée aux exigences de la directive Européenne sur les dispositifs médicaux, elle décrit un processus permettant aux fabricants de gérer les risques associés à un dispositif médical.
Cet article est une présentation générale de la norme, en 3 points:
- Utilité de la norme en vue de l’obtention du marquage CE d’un DM
- Les grands principes de l’ISO14971 pour la gestion des risques
- Place de la gestion des risques dans le cycle de vie du dispositif médical
À mettre en place une gestion efficace des risques induits par un dispositif médical, une activité imposée par la réglementation Européenne.
L’ISO 14971 est-elle obligatoire ?
Non, mais elle est reconnue comme moyen de réponse à diverses exigences Européenne et est systématiquement mise en œuvre par les fabricants de dispositifs médicaux.
Qu’est-ce qu’un risque ?
Au sens de la norme, un risque combine la probabilité d’occurrence et la gravité d’un dommage à une personne, ces deux aspects sont utilisés pour évaluer et maîtriser les risques.
Quelles sont les grandes lignes pour gérer les risques ?
Il faudra, du début de la conception à la fin de vie du produit, documenter un dossier de gestion des risques et faire vivre une analyse des risques, visant à identifier, évaluer et maîtriser chaque risque.
Importance de la gestion des risques en contexte “dispositif médical”
Gestion des risques et directive 93/42/CEE relative aux dispositifs médicaux
Pour rappel : le règlement (UE) 2017/745, appliqué dans la réglementation française, encadre les modalités de certification CE des dispositifs médicaux, il définit les différentes procédures de marquage CE, applicables en fonction de la classe des dispositifs médicaux.
Point commun de toutes ces procédures : la gestion d’une documentation technique incluant les résultats de l’activité de gestion des risques :
La documentation technique (…) comprend en particulier: (…) les résultats de l’analyse des risques
La notion de risque est également très présente dans les exigences générales du règlement, une recherche du terme “risque“met en évidence l’importance de cette notion qui est reprise une cinquantaine de fois !
Vous l’aurez compris : mettre en place une activité de gestion des risques est impératif pour pouvoir “marquer CE” vos produits.
Les normes harmonisées et la gestion des risques
Les autres normes harmonisées font très souvent référence à la gestion des risques, citons l’EN 60601-1 (exigences pour la sécurité et les performances des dispositifs électromédicaux) la conformité à de très nombreuses exigences sera démontrée via le dossier de gestion des risques, avec cette phrase récurrente :
La conformité est vérifiée par inspection du dossier de gestion des risques
La norme fixant les exigences sur les systèmes de management de la qualité est contexte DM (ISO13485) appel également la gestion des risques, c’est toujours le cas dans la prochaine révision, la 13485:2016 est explicite :
L’organisme doit documenter un ou plusieurs processus relatifs à la gestion des risques tout au long de la réalisation du produit
Note: Pour obtenir des informations relatives à la gestion des risques, se référer à d’autres documents, par exemple l’ISO 14971.
La gestion des risques selon l’ISO 14971
Qu’est-ce qu’un risque ?
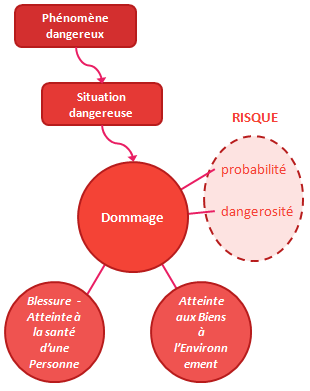
L’ISO14971 introduit plusieurs notions permettant de comprendre la notion de risque :
- Tout part d’un phénomène dangereux, souvent une défaillance du dispositif ou un mésusage.
- Sous certaines conditions peut naître une situation dangereuse : des personnes, des biens ou l’environnement sont alors exposés au phénomène dangereux.
- Cette situation dangereuse peut se traduire par des dommages : blessure ou atteinte à la santé, à l’environnement ou aux biens.
- Ces dommages sont caractérisés par leur probabilité d’occurrence et leur gravité : c’est cette combinaison qui définit la notion de risque.
Comment gérer les risques ?
Le processus de gestion des risques décrit dans l’ISO 14971 s’articule autour de plusieurs étapes, répétables jusqu’à maitrise totale des risques :
- Identification des risques : il s’agit d’imaginer tous les risques qui peuvent découler de l’utilisation du dispositif médical, on s’intéresse aux phénomènes dangereux et aux situations dangereuses induites.
- Évaluation des risques : les risques identifiés sont pondérés, selon leur probabilité et leur gravité, ceci permet d’exprimer la criticité de chaque risque. L’analyse de la criticité permet de statuer sur l’acceptabilité du risque, en tenant compte du rapport bénéfice-risque.
- Maîtrise du risque : un risque non accepté doit être maîtrisé : il est éliminé ou réduit par modification de la conception, ajout de fonction de sécurité, information de l’utilisateur…
- Évaluation du risque résiduel : les moyens de maîtrise du risque mis en place ont réduit le risque initial, mais subsiste un risque résiduel, il faut l’évaluer. En cas de non-acceptation, il faudra le maîtriser.
- Dernière étape : regarder si les moyens de réduction du risque non pas engendré de nouveaux risques ou modifier un risque déjà traité, auquel cas, il faudra évidemment recommencer le travail d’évaluation et de maîtrise.
Ces étapes sont le plus souvent formalisées dans une bonne grosse feuille Excel, souvent appelée “analyse des risques“.
Le dossier de gestion des risques
Le dossier de gestion des risques compile les informations utiles dans le cadre de la gestion des risques, c’est également un moyen de prouver que vos activités respectent les exigences de la norme (ce dossier sera soumis aux auditeurs, laboratoires d’essais et organismes notifiés). On y retrouve, entre autres, les informations nécessaires pour comprendre le dispositif et son contexte d’utilisation.
Quelques items clés du dossier de gestion des risques :
- Principe de fonctionnement du dispositif
- Liste des caractéristiques du DM liées à la sécurité
- Patients concernés (pathologies, schéma thérapeutique…)
- Bénéfice pour le patient
- Critère d’acceptabilité des risques
- Planification de la gestion des risques, responsabilité et autorités des différents acteurs
- …
Gérer les risques durant tout le cycle de vie du dispositif
Les activités de gestion des risques ne sont pas ponctuelles : elles sont initiées dès le début d’un projet et suivent le produit durant toute sa vie.
- En phase de conception et développement : l’analyse des risques va aboutir à la définition de moyens de réduction des risques, autant de nouvelles exigences sur le produit.
- En phase de production : les procédés et processus ayant un impact sur la sécurité du produit sont abordés sous l’angle de la gestion des risques, les moyens de maîtrise des risques pourront impacter la fabrication.
- Durant le stockage et le transport : tenez compte de l’impact des vibrations, des températures, de l’humidité, de la non-utilisation prolongée du dispositif…
- Après la mise sur le marché : vous avez mis en place les procédures de surveillance des produits appelées par la réglementation ? Très bien. Cette surveillance va très certainement remonter des situations dangereuses que vous n’aviez pas encore imaginées, vous voila bon pour faire une mise à jour de votre analyse des risques.
- Et même à la mise au rebut : où l’on pourra, par exemple, évaluer la dangerosité d’envoyer à l’incinérateur votre DM et sa bonne grosse batterie lithium-ion.
33 commentaires