EN ISO 13485:2016 – Les principales évolutions

La norme ISO 13485 vient d’évoluer, la version 2016 est une révision mineure de l’ISO 13485:2003, nous verrons ici les grandes lignes des changements, qui permettent une convergence vers les exigences réglementaires Européennes.
Cette révision devrait donc avoir un impact minime sur vos activités, il y a fort à parier que vous respectiez déjà bon nombre des nouvelles exigences.
(Les nouveautés seront étudiées dans le détail dans de futurs articles).
Historique : les révisions depuis 2003
Je vous propose d’observer les évolutions depuis 2003, tant pour la norme ISO 13485 (version internationale) que pour l’EN ISO 13485 (version Européenne) qui complète l’ISO avec des annexes Z, les exigences étant strictement les mêmes dans les deux versions.
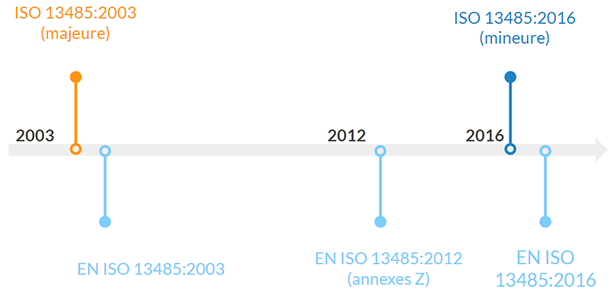
- La dernière révision de l’ISO 13485 date de 2003, c’était alors une révision majeure
- La version EN a connu une révision en 2012, portant uniquement sur les annexes Z (liens avec les exigences des directives sur les DM, DMIA et DMDIV)
- La version ISO 13485:2016 est sortie en mars 2016, l’EN ISO 13485:2016 a suivi en avril
- A noter que l’EN ISO 13485:2016 n’est pas dans la dernière liste des normes harmonisées à la 93/42/CEE, probablement pour laisser le temps aux organismes notifiés de former leurs auditeurs.
Pour info, les normes sont systématiquement étudiées tous les 5 ans par les comités techniques, suite à quoi une norme peut être confirmée (pas de changements), amendée (légers changements dans un amendement), révisée (c’est le cas de la 13485) ou annulée.
Des nouvelles définitions pour des nouvelles exigences
Les définitions introduites sont très révélatrices des changements de la norme, on peut les regrouper par familles:
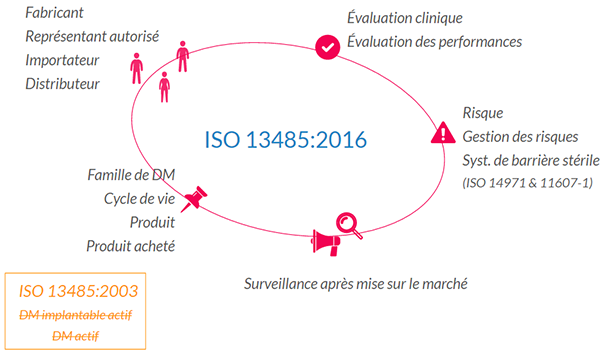
Les acteurs autour du DM
- Fabricant: responsable de la conception et/ou de la fabrication (notez le “ou” un fabricant peut donc ne rien fabriquer du tout).
- Représentant Autorisé: le mandataire, mandaté par le fabricant pour agir dans le cadre des taches réglementaires. Utile à un fabricant en dehors de la juridiction.
- Importateur: rends disponible le produit d’un fabricant hors pays/juridiction.
- Distributeur: rends disponible le dispositif médical au près de l’utilisateur final.
Ces définitions seront utilisées dans le texte de la norme qui vise à une meilleure implication de ces acteurs pour assurer la sécurité et les performances des DM, tout au long du cycle de vie.
Des évaluations précisées
- Évaluation clinique : étude des données cliniques pour démontrer la sécurité clinique et les performances d’un DM.
- Évaluation des performances : ne concerne – curieusement – que les DIV.
À part pour préciser que ces évaluations ne constituent pas une première mise sur le marché, ces définitions n’ont strictement aucun impact.
Définitions issues d’autres normes
- L’ISO 14971 (gestion des risques) donne les définitions de risque et de gestion des risques
- L’ISO 11607-1 celle de système de barrière stérile
Les barrières stériles sont évoquées dans le 7.5 “Production et prestation de service“, avec des exigences en termes de validation.
La notion de risque est beaucoup plus mise en avant, avec une philosophie: proportionner le travail en fonction des risques associés.
Surveillance après mise sur le marché
En vue de répondre aux exigences réglementaires applicables la surveillance après commercialisation vise à recueillir et à analyser les données “réelles” concernant la sécurité et les performances.
D’autres définitions plus générales
- Famille de DM: un groupe de dispositif aux caractéristiques similaires. L’idée est de gérer en une fois tout un groupe (pour le dossier de conception et le dossier du DM).
- Cycle de vie: de la conception à la mise au rebut, la norme concerne tout le cycle de vie.
- Produit et produit acheté: RAS
Principales nouveautés de l’ISO 13485:2016
Approche par les risques
L’ISO 13485:2016 demande de mettre en œuvre des actions proportionnées aux risques, l’approche fondée sur les risques est exigée en 4.1 :
“L’organisme doit appliquer une approche fondée sur les risques en ce qui concerne la maîtrise des processus appropriés nécessaires au SMQ”
Cette notion revient très souvent dans les exigences: validation des logiciels utilisés pour le SMQ, vérification de l’efficacité des formations, vérification du produit acheté…
Une norme à des fins réglementaires
Le titre de la norme à beau être explicite (“à des fins réglementaires“) l’obligation de tenir compte des exigences réglementaires est rappelée un peu partout, une bonne vingtaine de fois, notamment pour les exigences à prendre en compte pour le SMQ, la maitrise des informations médicales à caractère confidentiel, la définition des exigences sur le produit (en plus de celles du client), les processus de communication à mettre en place (avec les autorités réglementaires), le traitement du non-respect des spécifications d’achats…
En principe rien de nouveau: norme ou pas vous devez respecter la réglementation.
De l’importance des enregistrements
Ici aussi la norme bégaie, tout est dit en 4.1 (avec en prime un rappel sur les exigences réglementaires):
“[…]établir et conserver les enregistrements nécessaires pour démontrer la conformité à la présente Norme internationale et la conformité aux exigences réglementaires applicables”
Cette exigence est répétée absolument partout: éléments de sortie des revues, enregistrement des vérifications et des validations, achats, prestations, maitrise des équipements…
En principe rien de nouveau: précisé ou pas vous devez conserver les enregistrements nécessaires pour prouver la conformité de vos activités.
Il y a une vie après la mise sur le marché
Tendance forte (coucou PIP), le suivi après commercialisation est de plus en plus contrôlé, le futur règlement sur les DM va d’ailleurs dans ce sens.
La norme y revient fréquemment:
- Traitement des réclamations en donnée d’entrée de la revue de direction.
- Communication avec les autorités réglementaires (on ne parle plus uniquement de communication avec les clients).
- Deux nouveautés en 8.2: “surveillance et mesurage” et “signalement aux autorités réglementaires”.
Nouvelles procédures exigées
À ajouter à la liste des procédures obligatoires de l’ISO 13485:2003.
- Pour encadrer la validation des applications logicielles utilisées dans le SMQ.
- Pour les revues de direction (et ce n’est pas forcément du luxe).
- Pour le transfert de conception (étape “passage de relais” entre la R&D et la production).
- Maitrise des modifications de la C&D.
- Traitement des réclamations.
En vrac
- L’évaluation de l’aptitude à l’utilisation est mise en avant (vive la norme IEC 62366-1).
- Il faut construire un “dossier du dispositif médical” ainsi qu’un “dossier de conception et de développement” (rien d’effrayant pour qui connait les dossiers techniques de marquage CE).
- Nouvelles exigences autour de la maîtrise de la contamination.
- Distinguo des non-conformités détectées avant ou après la livraison.
- Exigences autour des retouches.
- L’UDI (identification unique) est évoqué, mais il faudra attendre que la réglementation l’encadre.
Conclusion
C’est une révision mineure, qui converge vers les exigences réglementaires déjà en en place, la transition ne devrait pas être trop douloureuse.
[Mise à jour de janvier 2019] L’alignement de la 13485 sur la structure HLS, qui doit simplifier la vie des organismes ayant une double certification ISO 9001:2015 et ISO 13485, est vraissemblablement repoussé à 2024, le temps de digérer le règlement.
12 commentaires