PR NF EN 62366-1:2014

Un projet de norme vient d’être soumis à enquête publique, il s’agit d’une évolution de l’EN 62366:2008, norme harmonisée au titre de la directive 93/42/CEE sur le DM, maintenant divisée en 2 parties, la seconde étant un guide d’application dont le draft n’a pas encore été publié.
Cette norme vise à prévenir les dangers dus aux erreurs d’utilisation d’un dispositif médical. Sa mise en application doit permettre une utilisation intuitive des dispositifs, ce qui est critique à l’heure où le patient devient de plus en plus son propre soignant, on pense aux dispositifs utilisés au domicile.
Cette nouvelle révision renforce les liens avec l’EN ISO 14971:2012 concernant la gestion des risques.
Utilisation normale, anormale et erreur d’utilisation
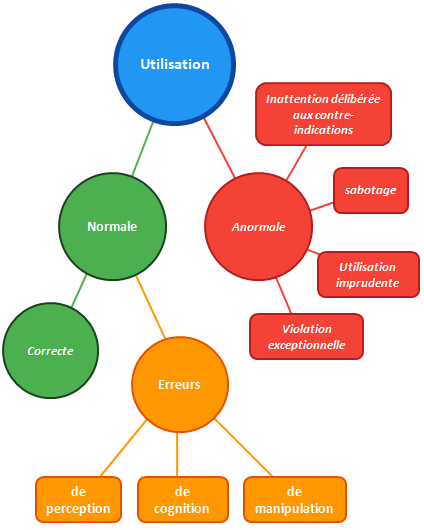
L’ingénierie de l’aptitude à l’utilisation ne concerne que l’utilisation normale d’un dispositif.
En effet, l’utilisation anormale ne peut être sécurisée par le fabricant, c’est notamment le cas lors :
- d’une violation exceptionnelle (utilisation complètement inappropriée du dispositif);
- d’une inattention délibérée aux contre-indications (l’utilisateur ne tient pas compte des informations relatives à la sécurité);
- d’une utilisation imprudente (sans tenir compte des phénomènes dangereux);
- d’un sabotage.
Dans de tels cas l’impuissance du fabricant est évidente.
L’utilisation normale se fait conformément à l’emploi prévu, tant sur l’utilisation du DM que sur les conditions (formation effectuée, instructions d’utilisation prises en comptes…). Mais même dans ce cas une utilisation incorrecte peut survenir, en cas d’erreur :
- de perception (ex : alarme non remarquée)
- de cognition (ex : valeur numérique mal interprétée)
- de manipulation (ex : commande mal actionnée)
Le but de l’IEC 62366-1 est d’éviter ces erreurs, en les identifiant et en les maîtrisant.
Place du processus d’ingénierie de l’aptitude à l’utilisation et intérêt vis-à-vis de la directive Européenne sur les dispositifs médicaux
Le mot est lâché : il faudra mettre en place un processus pour l’aptitude à l’utilisation.
Un organisme qui a mis en place un système de management de la qualité selon l’EN ISO 13485:2012 inclura l’aptitude à l’utilisation au processus de réalisation du produit (§7).
Le processus d’aptitude à l’utilisation va mettre en évidence des risques liés à l’utilisation, ces risques seront à traiter dans le processus de gestion des risques, selon l’EN ISO 14971:2012.
Traiter l’aptitude à l’utilisation permet de répondre à des exigences essentielles définies dans la directive 93/42/CEE.
La version 62366:2008 était harmonisées à la directive, avec une annexe ZZ précisant :
La conformité avec cette norme constitue une méthode de conformité avec les exigences essentielles spécifiées des Directives concernées.
La 62366-1:2014 n’a pas encore une telle annexe, mais on peut penser qu’elle couvre des exigences telles que :
(…) réduire, dans toute la mesure du possible, le risque d’une erreur d’utilisation due aux caractéristiques ergonomiques du dispositif (…)
La conception doit permettre une manipulation facile (…)
(vous pouvez faire une recherche parmi les différentes exigences via la webapp eqms.io, rubrique exigences essentielles 93/42/CEE).
Vue globale du processus d’ingénierie de l’aptitude à l’utilisation
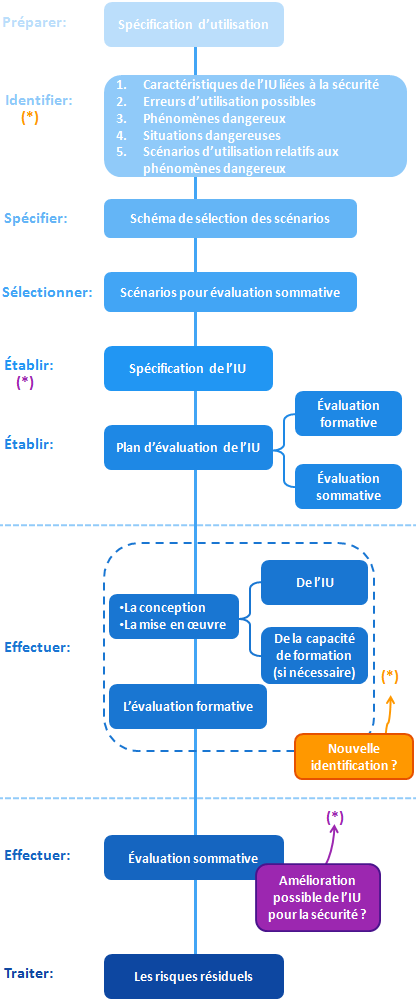
Trois grands temps ressortent de ce processus :
- Travail en amont de la réalisation de l’interface utilisateur (IU)
- Conception et développement de l’IU
- Évaluation de l’IU et traitement des risques résiduels
Travail en amont de la conception
Il s’agit de détailler, dans le dossier d’aptitude à l’utilisation, l’utilisation du dispositif et les risques associés.
Spécification de l’utilisation
Cette spécification complète celle préparée pour la gestion des risques, le fabricant y précise ce qu’il a prévu concernant :
- L’indication médicale
- La population de patients visée
- La partie du corps concernée
- L’environnent d’utilisation
- Le profil d’utilisateur
- Le principe de fonctionnement du DM
Identification des risques liés à l’utilisation
Le fabricant doit identifier les items suivants, dans l’ordre indiqué (les données de sortie d’une étape étant les données d’entrée de la suivante) :
- Caractéristiques de l’IU liées à la sécurité. En prenant en compte la sécurité du patient, du personnel soignant, de l’environnement, …
- Erreurs possibles d’utilisation de l’interface utilisateur
- Phénomènes dangereux qui en découlent (les sources potentielles de dommage)
- Situations dangereuses (où il y a une exposition aux phénomènes dangereux)
- Scénarios d’utilisation associés à ces phénomènes et situations (un scénario entre dans le cadre de la spécification d’utilisation et découpe les actions de l’utilisateur en taches, en mettant en regard de chaque tache les réponses du dispositif).
L’identification des phénomènes dangereux et des situations dangereuses n’est pas spécifique à la 62366-1, c’est une étape du processus de gestion des risques. Le lien IEC 62366-1 / ISO 14971 est évident.
Sélection des scénarios
Les scénarios identifiés vont être très nombreux et le traitement de chaque scénario est inenvisageable.
Il faut donc effectuer une sélection des scénarios qui seront ensuite utilisés pour l’évaluation sommative.
Cette sélection repose sur un schéma de sélection à spécifier.
Développement de l’interface utilisateur
Spécification
Il est demandé d’identifier :
- Les exigences pouvant être soumises aux essais.
- Si une documentation et/ou une formation sont nécessaires pour une utilisation sécurisée du dispositif.
Définition des plans d’évaluation
La norme repose sur 2 types d’évaluation de l’interface utilisateur (IU) :
- Évaluation formative : réalisée au fil du processus de la conception et du développement de l’IU. Il s’agit de valider durant le développement que l’interface est utilisable de manière correcte. Le fabricant peut par exemple tester en interne une partie de l’interface dès qu’elle est “présentable”. Cette évaluation va alimenter en permanence les données d’entrée de la conception, elle est susceptible de modifier la spécification de l’IU en révélant des nouvelles erreurs d’utilisation possibles, de nouvelles situations dangereuses…
- Évaluation sommative : effectuée une fois la conception du dispositif terminée, elle permet d’apporter des preuves tangibles sur l’utilisation du dispositif en toute sécurité.
Les plans d’évaluation contiennent les objectifs et les méthodes d’évaluation. En cas d’essais d’aptitude à l’utilisation il faudra documenter le profil des utilisateurs, l’environnent d’essai… autant de preuve que les essais sont représentatifs des spécifications de l’utilisation.
Conception et Mise en œuvre de l’IU
Le processus conception est développement suit son cours, en concevant l’interface utilisateur, en la mettant en œuvre et en réalisant l’évaluation formative au fur et à mesure des délivrables.
Si la spécification de l’IU le demande la capacité de formation est également conçue et mise en œuvre (créer des supports de formation et l’intendance nécessaire)
Évaluation sommative
L’évaluation sommative se fait selon les scénarios sélectionnés en respectant le plan en matière de profils d’utilisateurs, d’environnement d’essais, d’utilisation de documentation d’accompagnement, de formation.
Si cette évaluation met en évidence des améliorations possibles de l’IU pour la sécurité, il faudra reprendre le processus à l’étape adaptée.
11 commentaires