Marquage CE des dispositifs médicaux de classe I

Liste des actions que devra réaliser un fabricant pour pouvoir effectuer la déclaration CE de conformité d’un dispositif médical (DM) de classe I. Ce processus est décrit dans la directive Européenne 93/42/CEE relative aux DM, c’est une étape obligatoire avant la mise sur le marché d’un produit.
Quand avoir recours à la déclaration de conformité pour marquer CE un DM ?
Lorsqu’un fabricant souhaite mettre sur le marché un dispositif médical de classe I.
Qui intervient dans ce processus ?
Uniquement le fabricant, il n’y a pas de recours obligatoire à un organisme notifié.
Où sont définies les exigences ?
Dans l’annexe VII de la directive 93/42/CEE.
Que doit faire le fabricant ?
Constituer un dossier technique (créé dès le début du projet et tenu à jour tout au long de la vie du dispositif) et suivre les dispositifs après leur production, pour maîtriser les éventuels nouveaux risques.
Préambule
Avant de se lancer dans un processus de marquage CE le fabricant doit:
- Vérifier que le dispositif est bien un DM en mettant en regard l’usage revendiqué et la définition de dispositif médical donnée par la directive.
- Établir la classe du DM en fonction des règles décrites dans l’annexe IX de la 93/42/CEE.
La déclaration CE de conformité est un processus valable uniquement pour les DM de classe I, à noter que les dispositifs de classe I ayant une fonction de mesurage (Im) ou mis sur le marché à l’état stérile (Is) nécessitent des actions complémentaires.
Les DM de classe I ne sont pas censés présenter de risques importants, l’obtention du marquage CE est beaucoup plus simple que pour les DM de classes supérieures: c’est uniquement le fabricant qui a la responsabilité du marquage CE, il n’y a donc pas de recours à un organisme notifié. On parle d’auto-déclaration. Ceci est expliqué en introduction de la directive:
“considérant que […] les procédures d’évaluation de conformité pour les dispositifs de la classe I peuvent être effectuées, en règle générale, sous la seule responsabilité des fabricants, vu le faible degré de vulnérabilité de ces produits […]”
et au point 5 de l’article 11:
“Pour les dispositifs de la classe I, autres que ceux sur mesure et ceux destinés à des investigations cliniques, le fabricant suit, pour apposer le marquage CE, la procédure visée à l’annexe VII et établit, avant la mise sur le marché du dispositif, la déclaration CE de conformité requise.”
Marquage CE d’un DM de classe I : vue générale
Le processus est présenté dans l’annexe VII de la directive 93/42/CEE, on distingue deux grands axes:
- Création de la documentation technique
- Mise en place de processus obligatoires
Documentation technique associée au DM
La documentation technique se construit durant 3 grandes étapes (ce découpage est un découpage pratique, non décrit dans la directive):
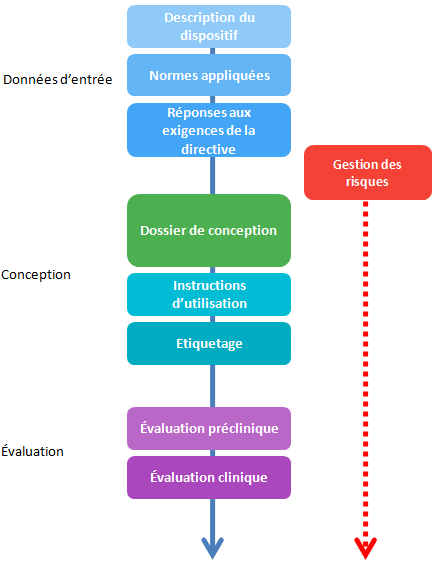
- Création des données d’entrée: où l’on décrit le produit et le contexte d’utilisation, les normes applicables ainsi que les moyens de répondre aux exigences essentielles de la directive.
- Conception: le travail de R&D à proprement parler durant lequel sera alimenté le dossier de conception. Les instructions d’utilisation et l’étiquetage sont généralement définis au fil du développement.
- Évaluation du dispositif, par des essais en laboratoire, des essais cliniques…
À noter que la gestion des risques sera initiée en début de conception et devra être mise à jour durant toute la vie du dispositif.
Processus obligatoires
Il n’est pas question de mettre ne place un système complet de management de la qualité, néanmoins des processus critiques sont à maîtriser: il s’agit principalement d’être en mesure de surveiller les produits sur le marché et de réagir efficacement en cas de risques.
Documentation technique en vue de la déclaration CE
Le contenu du dossier technique est résumé ci-dessous, la chronologie est mise en évidence:
Description du dispositif
La description concerne le dispositif mais aussi ses conditions d’utilisation. On y fera figurer:
- La finalité médicale
- Une explication du fonctionnement, avec des illustrations, des synoptiques…
- La liste des accessoires, des variantes, des autres dispositifs susceptibles d’êtres raccordés…
- Les caractéristiques techniques critiques
Remarque: Décrire le dispositif est également demandé pour gérer les risques, évaluer l’aptitude à l’utilisation, définir les essais à effectuer en laboratoire… On peut imaginer un unique dossier incluant de nombreux points, comme les profils de patients concernés, les conditions normales d’utilisations, les performances essentielles, la durée de vie prévue, les classifications utilisées pour évaluer la sécurité électrique… et ainsi simplifier la gestion documentaire. Il est tout à fait envisageable de regrouper ces informations dans le dossier de gestion des risques.
Normes appliquées
Le recours aux normes est un moyen reconnu pour répondre aux exigences sur les dispositifs médicaux, particulièrement avec les normes harmonisées à la directive 93/42/CEE.
Idéalement le dispositif est concerné par une “norme produit” (c’est par exemple le cas pour les oxymètres ou les lèves-personne) avec bien souvent des exigences sur les performances et les essais.
Dans tous les cas des “normes générales” sont applicables, elles peuvent concerner la sécurité électrique, les instructions d’utilisation ou encore la gestion des risques.
À noter que les normes sont rarement d’application obligatoire, ce sont des normes d’application volontaire. Cela peut être vu comme une contrainte, mais il faut comprendre que ces normes sont rédigées par des experts en consultation avec les différents acteurs concernés: tout un travail de réflexion que le fabricant n’aura pas à fournir, avec à la clé un produit plus sûr et un réel gain de temps.
Réponses aux exigences essentielles
L’idée est de balayer la liste des exigences essentielles, d’en vérifier l’applicabilité (en vous aidant de la description du dispositif) et de mettre en regard les moyens de mise en conformité (souvent via des normes).
Ces exigences sont définies dans l’annexe I de la directive 93/42/CEE.
Gestion des risques
Cela fait partie des exigences essentielles: il faut maîtriser les risques liés à l’utilisation du dispositif.
La norme harmonisée EN ISO 14971 “Application de la gestion des risques aux dispositifs médicaux” est un référentiel reconnu.
La gestion des risques sera initiée dès le début du développement et mise à jour en permanence.
Le fabricant tient à jour un dossier de gestion des risques et une analyse des risques regroupant l’identification, l’évaluation et les mesures de maîtrise des risques.
Dossier de conception
Cette documentation est très technique, elle doit permettre de comprendre comment fonctionne le DM. Elle inclut le cas échéant:
- Des plans d’assemblages
- Des schémas électriques, nomenclatures, CAO…
- Des documentations du logiciel selon l’IEC 62304
- Des notes de calculs
- …
Instructions d’utilisation et étiquetage
Là aussi il faut se reposer sur des normes harmonisées, EN 980 et EN 1041, ainsi que sur les résultats de la gestion des risques (cas typique des avertissements vis-à-vis des risques résiduels pourtant décriés dans l’annexe ZA de l’EN ISO 14971).
Évaluation pré-clinique
Il s’agit de vérifier que le dispositif:
- est conforme aux exigences fonctionnelles
- n’est pas dangereux
Une partie de ces vérifications est faite en interne, via des mesures, des tests, des simulations… une autre fait appel aux laboratoires d’essais, par exemple pour les essais de sécurité électrique selon l’EN 60601-1 ou de compatibilité électromagnétique selon l’EN 60601-1-2. Notez qu’un DM destiné à être utilisé au domicile du patient est également soumis à la norme EN 60601-1-11. Autant de spécificités pour la certification des dispositifs médicaux électriques.
Évaluation clinique
L’évaluation clinique peut nécessiter des investigations cliniques. La norme harmonisée EN ISO 14155 “Investigation clinique des dispositifs médicaux pour sujets humains” vous aidera à construire ces essais.
Dans de nombreux cas, pour des procédés maîtrisés, une revue de la littérature / de l’état de l’art peut suffire pour démontrer l’efficacité clinique.
Processus à mettre en place par le fabricant
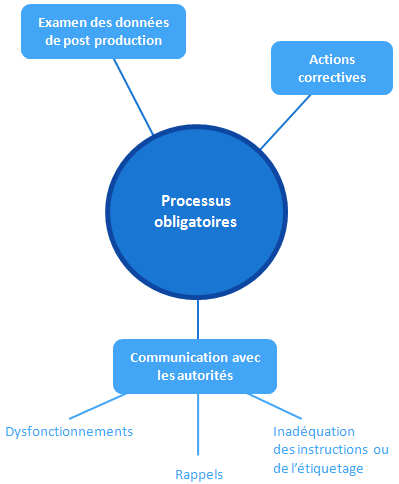
Le dossier technique démontre que le dispositif répond aux exigences de la directive, il faut maintenant s’assurer que cette conformité restera valable tout au long de la commercialisation et de l’utilisation du DM.
Le fabricant doit mettre en place les moyens de réponse à trois types d’exigences:
- être en mesure de collecter et de traiter les données acquises depuis la production (on pense aux informations liées à la sécurité: prise en compte d’un incident lié à l’utilisation du dispositif ou d’un dispositif comparable)
- pouvoir mettre en œuvre des actions correctives en réponse aux problèmes remontés
- assurer la communication avec les autorités compétentes (l’ANSM en France) en cas de dysfonctionnements, de rappels, d’inadéquation des instructions fournies ou du marquage, qui pourraient engendrer un risque non négligeable.
C’est au travers de procédures documentées que la conformité est établie.
Lien
Un guide de la commission Européenne à destination des fabricants de dispositifs médicaux de classe 1.
83 commentaires