Dossier : Emballages des dispositifs médicaux stériles
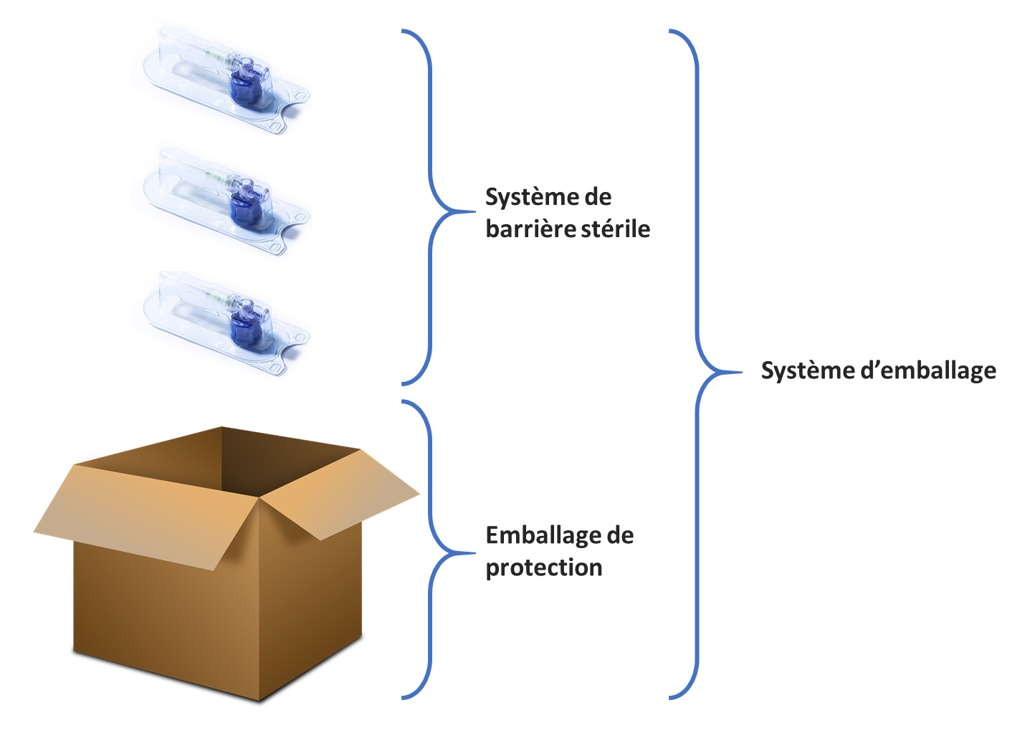
De nombreux dispositifs médicaux doivent être stérilisés afin que soit maitrisé le risque de contamination microbienne, c’est par exemple le cas de dispositifs invasifs, tels que les aiguilles de seringue ou les implants. Plusieurs procédés existent pour stériliser les DM mais encore faut-il que ces derniers restent stériles. Pour cela, il faut prévoir un emballage adéquat afin d’empêcher toute contaminations après stérilisation. La nature de cet emballage va dépendre de nombreux facteurs qui devront être pris en compte lors de la conception.
Ce dossier vous présente les fondamentaux pour la conception, le développement et la validation des systèmes d’emballage stérile.
Contexte
Règlementation applicable aux emballages de DM stériles
Le règlement 2017/745 aborde le sujet des dispositifs médicaux stériles à plusieurs reprises, nous retiendrons principalement ce qu’il dit dans l’Annexe I chapitre II.11 (exigences relatives à la conception et à la fabrication / Infection et contamination microbienne) :
« Les dispositifs livrés à l’état stérile sont conçus, fabriqués et conditionnés selon des procédures appropriées, pour garantir qu’ils sont stériles lors de leur mise sur le marché et qu’à moins que le conditionnement destiné à en préserver l’état stérile ne soit endommagé, ils restent stériles dans les conditions de transport et de stockage préconisées par le fabricant, jusqu’à ce que ce conditionnement soit ouvert au moment de l’utilisation…
Les dispositifs étiquetés comme étant stériles sont traités, fabriqués, conditionnés et stérilisés grâce à des méthodes appropriées et validées.
Les systèmes de conditionnement… lorsque ces dispositifs sont destinés à être stérilisés avant leur utilisation, réduisent au minimum le risque de contamination microbienne; le système de conditionnement est adapté à la méthode de stérilisation préconisée par le fabricant… »
Il appartient donc au fabricant de :
- Définir le type de conditionnement approprié au dispositif et au mode de stérilisation
- Réaliser la validation du procédé de conditionnement appliqué
- Maintenir cet état validé
Gestion des risques
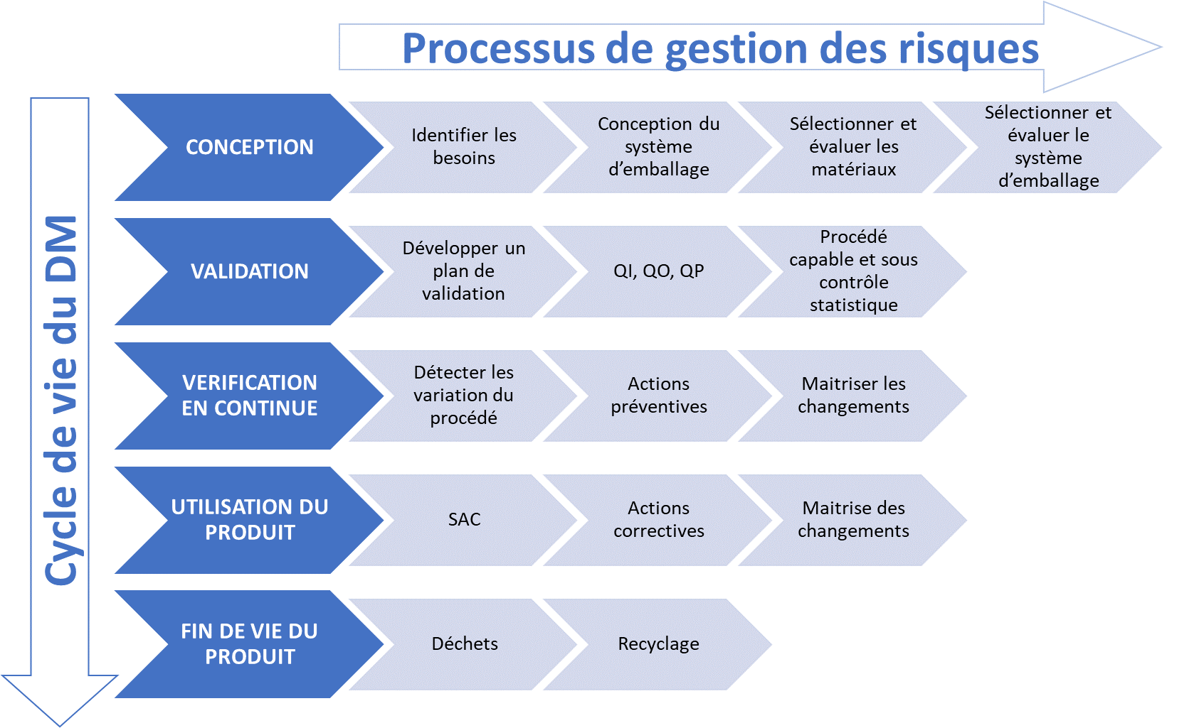
Concernant le conditionnement des DM (et comme pour le reste) il faut appliquer un processus de gestion des risques aux différentes étapes du cycle de vie du dispositif, tel que présenté ci-dessous :
Normes applicables
Les normes applicables (et harmonisées) sont l’ISO 11607-1 et l’ISO 11607-2, elles concernent :
- Les matériaux utilisés
- Les systèmes de barrière stérile (préformés ou non)
- Les systèmes d’emballage destinés à maintenir l’état de stérilité des dispositifs médicaux stérilisés au stade terminal jusqu’au point d’utilisation.
Elles seront appliquées par :
- L’industrie du dispositif médical (fabricant, sous-traitants, …)
- Les installations “de santé”
- Tout lieu où les dispositifs médicaux se trouvent dans des systèmes de barrière stérile et stérilisés.
Ces normes ne couvrent pas les dispositifs médicaux fabriqués de manière aseptique, c’est-à-dire ceux pour qui la stérilisation dans le conditionnement définitif n’est pas possible (exemple les seringues pré remplies).
Les principales nouveautés des révisions 2019 sont données ci-dessous.
Nouveautés de l’ISO 11607-1:2019
- Mise à jour des définitions
- Exigence d’évaluation de l’aptitude à l’utilisation (référence aux évaluations formatives et sommatives de l’IEC 62366-1).
- Exigence d’inspection de l’intégrité avant utilisation
- Exigences de revalidation
- Mise à jour de l’annexe B : suppression / ajout de méthodes d’essais
Nouveautés de l’ISO 11607-2:2019
- Mise à jour des définitions
- Plus de « paramètres critiques de procédé »
- Introduction du concept de « spécification de procédé »
Identifier les besoins, concevoir et développer
Principes
L’objectif de cette étape est d’obtenir un système d’emballage stérile permettant de présenter le produit d’une manière aseptique. C’est-à-dire :
- Laisser passer l’agent stérilisant, gaz, vapeur ou rayonnements ionisants, afin de détruire les bactéries enfermées à l’intérieur.
- Empêcher le passage des bactéries vivantes qui se trouve à l’extérieur de rentrer et de contaminer de nouveau l’intérieur du système de barrière stérile et le DM qui s’y trouve.
- Conserver cette « étanchéité » à la contamination malgré les agressions mécaniques, physiques et chimiques provoquées par le transport, le stockage et toutes les manipulations qu’il subira avant d’être utilisé.
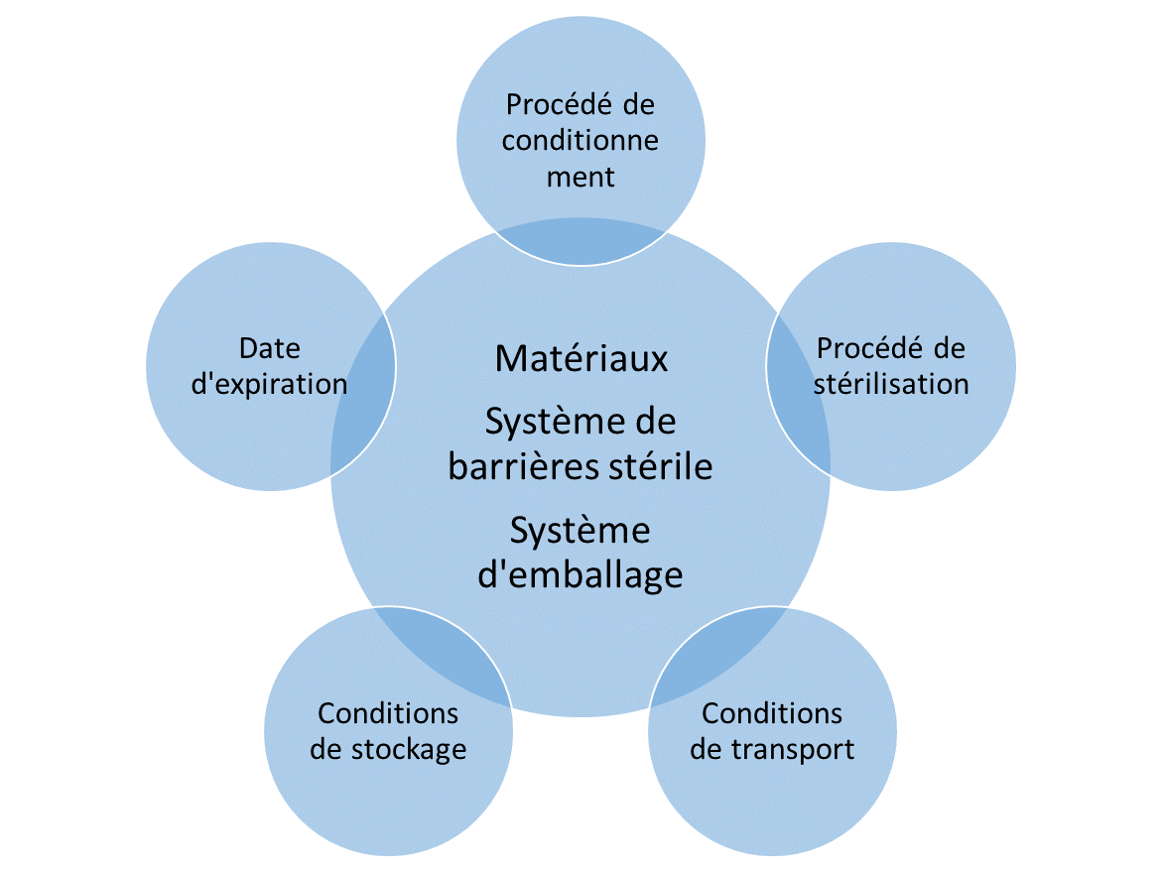
Les facteurs à prendre en compte lors de la conception sont nombreux. Chacun des éléments ci-dessous doit être déterminé afin que le résultat final réponde à l’objectif :
Référentiels applicables
- Norme ISO 13485 § 7.3 « conception et développement »
- Guide GHTF “Design Control Guidance For Medical Device Manufacturers”
Sélectionner et évaluer les matériaux
Principes
Pour sélectionner les matériaux appropriés, les éléments suivants doivent être évalués :
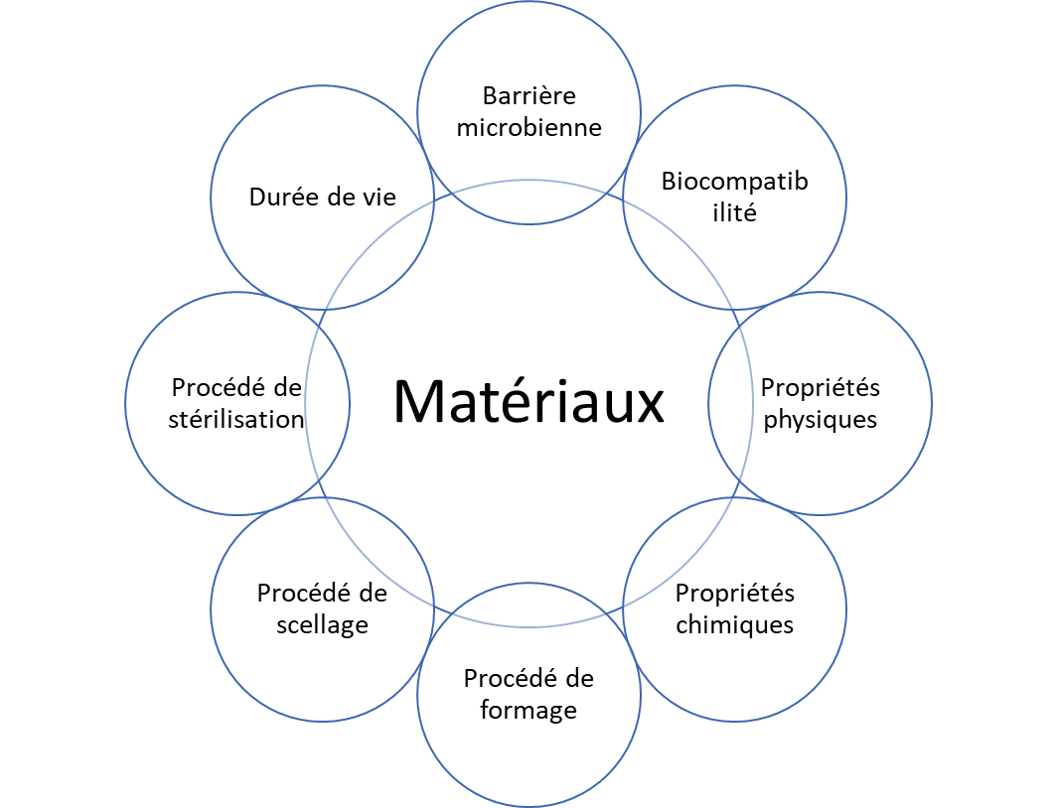
Il ne faut pas confondre l’évaluation du matériau avec l’évaluation de système de barrière stérile. La plupart du temps, l’évaluation du matériau est réalisée par le fournisseur et l’évaluation de la barrière stérile est faite par le fabricant (ou son sous-traitant). Le cas intermédiaire est celui du fournisseur de sachets préformés. Dans ce cas, 2 ou 3 soudures sont réalisées par le fournisseur du sachet et les dernières par le fabricant (ou son sous-traitant).
Pour les matériaux ou sachets achetés, il faut, à minima, vérifier leur conformité par examen des certificats qui doivent accompagner les produits. Des essais par échantillonnage peuvent également être réalisés afin de maitriser au mieux les risques relatifs aux matériaux.
Référentiels applicables
| Objectif général | Objectif détaillé | Normes applicables |
|---|---|---|
| Valider la perméabilité à l’air | permettre à l’agent stérilisant de traverser le matériau | ISO 5636-5 EN 868-2 |
| Vider l’efficacité de la barrière microbienne | empêcher l’agent contaminant de traverser le matériau | ASTM F1608 |
| Valider la biocompatibilité | démontrer que le matériau n’est pas toxique | ISO 10993-1 |
| Valider la tenue aux essais de vieillissement accéléré | démontrer que le matériau doit conserver ses caractéristiques durant toute sa durée de vie | ASTM F1980 EN 868-8 |
Concernant les différents procédés de stérilisations les normes suivantes peuvent être appliquées selon les cas pour démontrer la conformité des matériaux :
| Matériaux | Procédé de stérilisation | Norme applicable |
|---|---|---|
| Papier de grade médical non enduit d’adhésif | Vapeur | EN 868-3 |
| Gaz basse température | EN 868-6 | |
| Papier de grade médical enduit d’adhésif | Gaz basse température | EN 868-7 |
| Polyoléfine non enduit d’adhésif | Peroxyde d’hydrogène | EN 868-9 |
| Oxyde d’éthylène | EN 868-9 | |
| Polyoléfine enduit d’adhésif | Oxyde d’éthylène | EN 868-10 |
Même chose pour les sachets préformés ou gaines en rouleaux (système de barrière stérile en devenir) :
| Système de barrière stérile | Procédés de stérilisation | Normes applicables |
|---|---|---|
| Papier non enduit / film | Vapeur | EN 868-3, EN 868-5, EN ISO 11607-1 |
| Gaz basse température | EN 868-6, EN 868-5, EN ISO 11607-1 | |
| Papier enduit / film | Gaz basse température | EN 868-7, EN 868-5, EN ISO 11607-1 |
| Polyoléfine non enduit / film | Peroxyde d’hydrogène | EN 868-9, EN 868-5, EN ISO 11607-1 |
| Oxyde d’éthylène | EN 868-9, EN 868-5, EN ISO 11607-1 | |
| Polyoléfine enduit / film | Oxyde d’éthylène | EN 868-10, EN 868-5, EN ISO 11607-1 |
On constate ici qu’en plus des normes relatives aux matériaux, il faut également prendre en compte celles relatives au scellage et au système de barrière stérile puisque le fournisseur a réalisé certaines soudures (2 pour les rouleaux et 3 pour les gaines).
Évaluer le système de barrière stérile
Principes
Tous les essais décrits ci-dessous, Intégrité, résistance, barrière microbienne, doivent être réalisés après avoir subi l’ensemble des contraintes représentatives de la vie du dispositif :
- Stérilisation,
- Vieillissement (accéléré ou non),
- Transport
et ce, dans les conditions les plus défavorables :
- Paramètres des procédés de formage et de scellage les plus défavorables (limite haute et limite basses)
- Plusieurs expositions au procédé de stérilisation
Pour démontrer l’intégrité du système de barrière stérile il faut démontrer les propriétés de barrière stérile des matériaux ainsi que l’intégrité des scellages / fermetures.
Attention : certains essais réalisés sur les matériaux devront être refaits sur les produits finis (système de barrière stérile). En effet, le matériau peut avoir passé avec succès tous les tests nécessaires, cela ne permet pas de démontrer que le système de barrière stérile répond à toutes les exigences.
Référentiels applicables
Les méthodes recommandées dépendent du procédé de stérilisation et du type d’emballage :
Stérilisation à l’oxyde d’éthylène
| Vérification | Objectif | Type d’emballage | Référentiel |
|---|---|---|---|
| Intégrité du système de barrière stérile | empêcher l’agent contaminant de traverser le matériau ou les soudures / fermetures | Tyvek/ Blister rigide Poly / Poly |
Norme ASTM F2096 : bubble test |
| Résistance du système de barrière stérile | assurer que le système d’emballage résistera à toutes les agressions | Tyvek / Mylar Papier / Mylar Papier / Papier Sac à fenêtre Tyvek ou papier |
Norme EN 868-5 : Essais de traction Norme ASTM F88 : Essais de traction |
| Tyvek/ Blister rigide Poly / Poly |
Norme ASTM F1140 : Burst (éclatement) | ||
| Efficacité de la barrière microbienne du système de barrière stérile | empêcher l’agent contaminant de traverser le matériau et/ou les soudures / fermetures | Tyvek / Mylar Tyvek/ Blister rigide Papier / Mylar Papier / Papier Sac à fenêtre Tyvek ou papier |
Norme ASTM F1608 : Classification microbienne |
| Poly / Poly | Perméabilité à l’air |
Stérilisation par irradiation
| Vérification | Objectif | Type d’emballage | Référentiel |
|---|---|---|---|
| Intégrité du système de barrière stérile | empêcher l’agent contaminant de traverser le matériau ou les soudures / fermetures | Papier / Mylar Papier / Papier | Norme ASTM F1929 : dye penetration test |
| Nylon Tyvek / Mylar Tyvek/ Blister rigide Sac à fenêtre Tyvek ou papier Poly / Poly |
Norme ASTM F2096 : bubble test | ||
| Résistance du système de barrière stérile | assurer que le système d’emballage résistera à toutes les agressions | Papier / Mylar Papier / Papier | Norme EN 868-5 : Essais de traction Norme ASTM F88 : Essais de traction |
| Poly / Poly | Norme ASTM F1140 : Burst (éclatement) | ||
| Efficacité de la barrière microbienne du système de barrière stérile | empêcher l’agent contaminant de traverser le matériau et/ou les soudures / fermetures | Tyvek / Mylar Tyvek/ Blister rigide Papier / Mylar Papier / Papier Sac à fenêtre Tyvek ou papier |
Norme ASTM F1608 : classification microbienne |
| Poly / Poly | Perméabilité à l’air |
Stérilisation vapeur
| Vérification | Objectif | Type d’emballage | Référentiel |
|---|---|---|---|
| Intégrité du système de barrière stérile | empêcher l’agent contaminant de traverser le matériau ou les soudures / fermetures | Papier / Mylar Papier / Papier | Norme ASTM F1929 : dye penetration test |
| Résistance du système de barrière stérile | assurer que le système d’emballage résistera à toutes les agressions | Papier / Mylar Papier / Papier | Norme EN 868-5 : Essais de traction Norme ASTM F88 : Essais de traction |
| Efficacité de la barrière microbienne du système de barrière stérile | empêcher l’agent contaminant de traverser le matériau et/ou les soudures / fermetures | Papier / Mylar Papier / Papier | Norme ASTM F1608 : classification microbienne |
Résister aux transports
Principes
Les circuits logistiques dépendent :
- des modes de transport
- des destinations
- des types de produits
- des acteurs impliqués
Ces circuits de distribution imposent des contraintes au produit:
- Cahot lors du transport
- Manutention,
- Chargement(s) et déchargement
- Conditions de stockage plus ou moins maitrisées
L’objectif d’une validation de transport est de simuler, par des essais normalisés, ces contraintes mécaniques ou climatiques sur le produit emballé, représentatif de ce qu’il sera en condition réelle, et de vérifier si le produit a conservé son aptitude à l’emploi. L’emballage doit également garder ces caractéristique pour être manipulable.
Référentiels applicables
Les principaux programmes d’essais sont décrits dans les référentiels suivants :
- NF H 00-060
- NF EN 15552,
- ASTM D 4169
- le référentiel technique de l’International Safety Transit Association (ISTA).
- l’ISO 4180
Des programmes personnalisés peuvent également être développés selon les contraintes particulières spécifiques au produit.
Exemple de programme avec la norme NF H 00-060 « Emballages d’expédition complets et pleins. Programmes d’essais »
Dans cette norme, un programme d’essais est élaboré selon les critères suivants:
- Séquence d’essais (A à E) choisie en fonction des dimension, masse et type d’emballage.
- Sévérité mécanique (1 à 9) choisi en fonction du type de transport, du circuit de distribution, de la hauteur de gerbage …
- Condition climatique (D, E, Y, …) choisi en fonction du matériaux de l’emballage et des destinations.
Gérer la date d’expiration / le vieillissement du dispositif
Principes
Le système de barrière stérile doit pouvoir démontrer que ses performances sont maintenues jusqu’à l’utilisation. Pour cela, un essai de stabilité doit être effectué à l’aide d’un procédé de vieillissement en temps réel.
Cependant, l’essai de stabilité à l’aide de procédés de vieillissement accéléré est considéré comme une preuve suffisante de date d’expiration revendiquée tant que les données issues des études de vieillissement en temps réel ne sont pas disponibles. Les essais de vieillissement accéléré et en temps réel doivent être lancés en simultané.
Référentiel applicable
L’essai de vieillissement peut être réalisé selon l’ASTM 1980, norme basée sur la loi d’Arrhenius sur la vitesse de réaction :
Une élévation de la température de 10°C doublera la vitesse d’une réaction chimique.
Par exemple, un système de barrière stérile soumis à une température de 55°C pendant 5,3 semaines sera considéré comme équivalent au même produit soumis à une température de 22°C pendant 1 an.
Concevoir les informations fournies à l’utilisateur
Principes
Les informations suivantes doivent être fournies avec le matériau, le système de barrière stérile préformé ou le système de barrière stérile :
- le type, la taille ou le grade ;
- le numéro de lot ou un autre moyen permettant le suivi de l’historique de fabrication ;
- le(s) procédé(s) de stérilisation prévu(s) ;
- la date d’expiration, le cas échéant ;
- toute condition de stockage spécifique, le cas échéant ;
- toute restriction connue relative à la manipulation ou l’utilisation (conditions environnementales par exemple), le cas échéant ;
- pour les matériaux réutilisables et/ou les systèmes de barrière stérile préformés, la fréquence et la nature de l’entretien.
Lorsque des réglementations nationales ou régionales requièrent des informations supplémentaires sur les systèmes de barrière stérile commercialisés sur le marché de la santé, celles-ci doivent être fournies.
Référentiel applicable
La norme ISO 11607-1 est applicable, la révision 2019 précise que le mode d’emploi doit inclure des instructions pour l’inspection visuelle des ruptures de l’intégrité de l’emballage avant utilisation.
Valider les procédés
Selon l’ISO 11607-2, la validation des procédés est réalisée selon 3 niveaux de qualification QI, QO et QP :
- Qualification de l’installation
- Qualification Opérationnelle
- Qualification de performance
Cette méthode sera décrite dans un prochain article.
Réaliser la Surveillance Après Commercialisation
La phase de surveillance après commercialisation permettra de collecter des informations relatives à la vie du dispositif en conditions réelles.
Ces informations et leur analyse pourront amener le fabricant à revoir tout ou partie de son système d’emballage ou des procédés de conditionnement.
8 commentaires