Procédures d’évaluation de la conformité – directive 93/42/CEE
Voir également le guide actualisé du marquage CE des dispositifs médicaux.
Présentation des procédures d’évaluation de la conformité des dispositifs médicaux (DM) définies dans la directive Européenne 93/42/CEE. Ces procédures permettent aux fabricants de démontrer le respect des exigences de la directive, matérialisé par le marquage CE du dispositif et la déclaration CE de conformité faite par le fabricant.
Un préambule permet de comprendre les concepts utilisés dans la directive, les procédures sont ensuite présentées en fonction de la classe du dispositif médical.
Notez que chaque dispositif a ses spécificités pour le marquage CE, notamment les dispositifs électriques.
Qu’est-ce que le “marquage CE” d’un dispositif médical ?
Le marquage CE traduit le fait qu’un dispositif est conforme aux exigences applicables en Europe et qu’il a été évalué selon les procédures prévues. Lorsqu’applicable la marque CE est suivie du numéro d’identification de l’organisme notifié qui est intervenu pour établir la conformité.
Où sont définies ces procédures d’évaluation ?
Dans l’article 11 de la 93/42/CEE “Évaluation de la conformité”, complété par les annexes II à VII.
Comment choisir la procédure à appliquer ?
Il faut d’abord déterminer la classe du DM pour connaitre les procédures applicables. Le fabricant choisit ensuite le moyen le mieux adapté aux activités de son entreprise.
Sur quoi porte l’évaluation de la conformité ?
La mise en conformité demande, à minima, de gérer un dossier technique (dossier d’évaluation) et de mettre en place certaines procédures. Un organisme notifié devra intervenir pour les dispositifs autres que de classe I.
Quel est le rôle de l’organisme notifié ?
L’ON peut, en fonction de la classe du DM et de la procédure choisie: évaluer la conception du dispositif, évaluer le système qualité (totalement ou en partie), effectuer le contrôle final des produits.
Préambule
Classe d’un dispositif médical
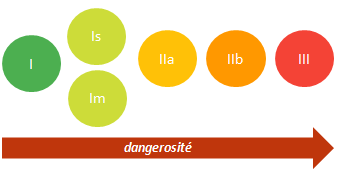
La classe caractérise la dangerosité potentielle d’un DM, l’annexe IX de la directive 93/42 définit les règles de classification.
Un article du blog présente les critères de classification d’un DM, vous pouvez rapidement définir la classe via ce questionnaire.
4 classes sont utilisées: I, IIa, IIb et III. Les dispositifs de classe I intégrant une fonction de mesurage (Im) ou vendus à l’état stérile (Is) ont des exigences supplémentaires.
Activité critiques du fabricant
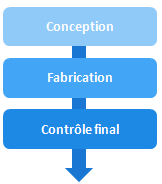
Les procédures d’évaluation distinguent 3 activités:
- Conception (la phase de R&D)
- Fabrication (la phase de production)
- Contrôle final (la phase de vérification de la conformité du produit fabriqué)
En fonction de la classe et de la procédure choisie, tout ou partie de ces activités seront contrôlées par un organisme notifié.
Documentation technique et processus obligatoires
Indépendamment de la procédure choisie, le fabricant devra:
- Gérer une documentation technique (appelé aussi dossier de conformité) contenant une description du dispositif, les moyens mis en œuvre pour répondre aux exigences de la directive, des détails sur la conception, l’analyse des risques, les rapports d’essais, l’évaluation clinique…
- Mettre en place des processus obligatoires pour garantir que les dispositifs mis sur le marché seront suivis et qu’en cas de problème des actions adaptées seront mises en œuvre.
Mise en place d’une assurance qualité
L’expression “assurance qualité” est à prendre au pied de la lettre: la qualité des activités du fabricant est garantie par la mise en place d’un système de management de la qualité (SMQ).
Pour ce faire, le fabricant applique les exigences d’une norme de SMQ et fait certifier son système par un organisme notifié.
Dans l’industrie du dispositif médical c’est la norme harmonisée EN ISO 13485 “Dispositifs médicaux — Systèmes de management de la qualité — Exigences à des fins réglementaires” qui est reconnu comme moyen de satisfaire aux exigences de la directive.
Recours à un organisme notifié
Un organisme notifié (O.N.) a pouvoir de valider l’assurance qualité du fabricant. Ces organismes sont désignés par les autorités compétentes, également en charge de leur surveillance.
Les ON peuvent également réaliser le contrôle final d’un DM ou délivrer un certificat de conformité pour un type de produit.
Le fabricant est libre de travailler avec l’organisme de son choix, la liste des ON pour la 93/42/CEE est disponible sur europa.eu.
Évaluation de la conformité des DM de classe I, Im et Is
Auto-déclaration CE de conformité (classe I)
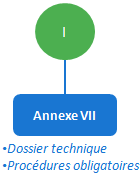
C’est le cas le plus simple, défini dans l’annexe VII, le fabricant est seul responsable de la déclaration de conformité du produit, il devra gérer le dossier technique et mettre en place les procédures obligatoires. (voir cet article pour plus de détails).
Remarque: ce processus est pensé pour les DM dont l’utilisation n’est pas dangereuse, mais les règles de classification n’étant pas infaillibles il y a régulièrement des décès avec des dispositifs de classe I ! Je pense aux dispositifs permettant de déplacer des personnes handicapées : une défaillance peut entraîner une chute qui, chez les plus faibles, peut causer la mort.
Mesure complémentaire pour les classes Im et Is
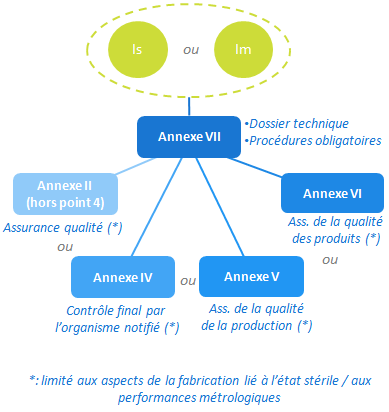
Les dispositifs de classe I intégrant une fonction de mesurage ou mis sur le marché à l’état stérile demandent une assurance de la qualité de la production, limitée aux aspects “métrologie” ou “stérile”.
L’annexe VII est donc à compléter par (au choix) l’annexe II, IV, V ou VI.
Évaluation de la conformité des DM de classe IIa et IIb
Système complet d’assurance qualité (IIa et IIb)
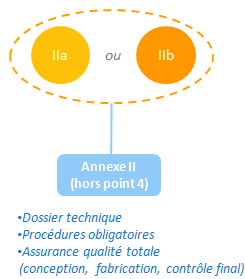
La procédure définit dans l’annexe II hors point 4 est commune aux DM de classe IIa et IIb.
Comme pour chaque procédure le fabricant génère un dossier technique et met en place les processus obligatoire.
Il met également en place un système complet d’assurance qualité, contrôlé par un organisme notifié.
Remarque: Ce choix est relativement lourd à mettre en œuvre (une certification 13485 demande en gros 1 an de travail) mais il est également souple: l’organisme notifié ne contrôle pas le produit mais les activités du fabricant, une fois celles-ci bien encadrées il est relativement aisé de certifier de nouveaux produits, pourvus qu’ils soient dans le champ d’application précisé sur le certificat d’assurance qualité délivré à l’entreprise.
Auto-déclaration de conformité et mesures complémentaires (IIa)
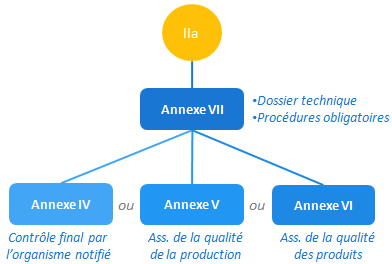
Tout comme pour la classe I, un fabricant de DM de classe IIa peut faire une auto-déclaration conformité (annexe VII) à compléter avec l’une des 3 procédures ci-dessous:
- Contrôle final réalisé par l’organisme notifié: selon l’annexe IV, on parle de vérification CE. Le contrôle pouvant être effectué sur chaque produit ou par prélèvement statistique. Il permet de vérifier la bonne mise en œuvre des mesures décrites dans le dossier technique.
- Assurance qualité de la production (annexe V).
- Assurance de la qualité des produits (annexe VI): le fabricant fait certifier ses activités de contrôle final.
Examen CE de type et mesures complémentaires (IIb)
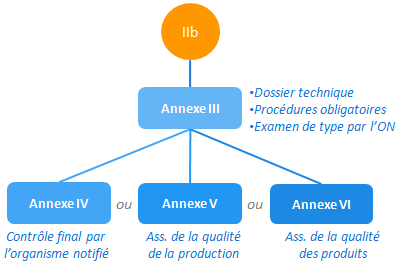
L’examen CE de type est prévu par l’annexe III:
- le dossier technique est contrôlé par l’organisme notifié.
- un échantillon représentatif de la production est soumis à l’ON qui vérifie sa conformité au dossier technique et effectue les essais nécessaires.
En cas de succès l’ON délivre un certificat d’examen de type, valable 5 ans.
L’annexe III est à compléter, au choix, par les annexes IV, V ou VI.
Évaluation de la conformité des DM de classe III
Système complet d’assurance qualité et examen de la conception
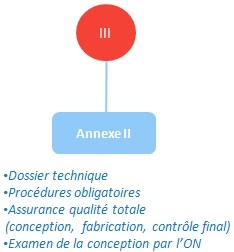
Pour les DM de classe III l’annexe II s’applique entièrement: en plus d’un SMQ complet la conception est examinée par l’organisme notifié qui délivre un certificat d’examen CE de la conception, d’une durée limitée à 5 ans.
Remarque: Cette procédure est la plus exigeante, elle est justifiée par les risques liés aux DM de classe III (on retrouve dans cette classe des dispositifs implantables, de maintien de la vie, chirurgicaux,… qui peuvent provoquer le décès du patient en cas de défaillance / de mauvaise conception).
Examen CE de type et mesures complémentaires
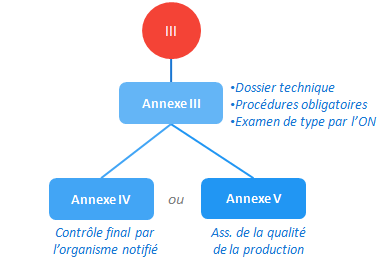
Un cas comparable à la classe IIb mais ici l’assurance de la qualité des produits n’est pas possible, on a donc une annexe III (examen CE de type) à compléter par l’annexe IV (contrôle final réalisé par l’ON) ou par l’annexe V (assurance qualité de la production).
Cadeau bonus: les procédures applicables en fonction de la classe en une image
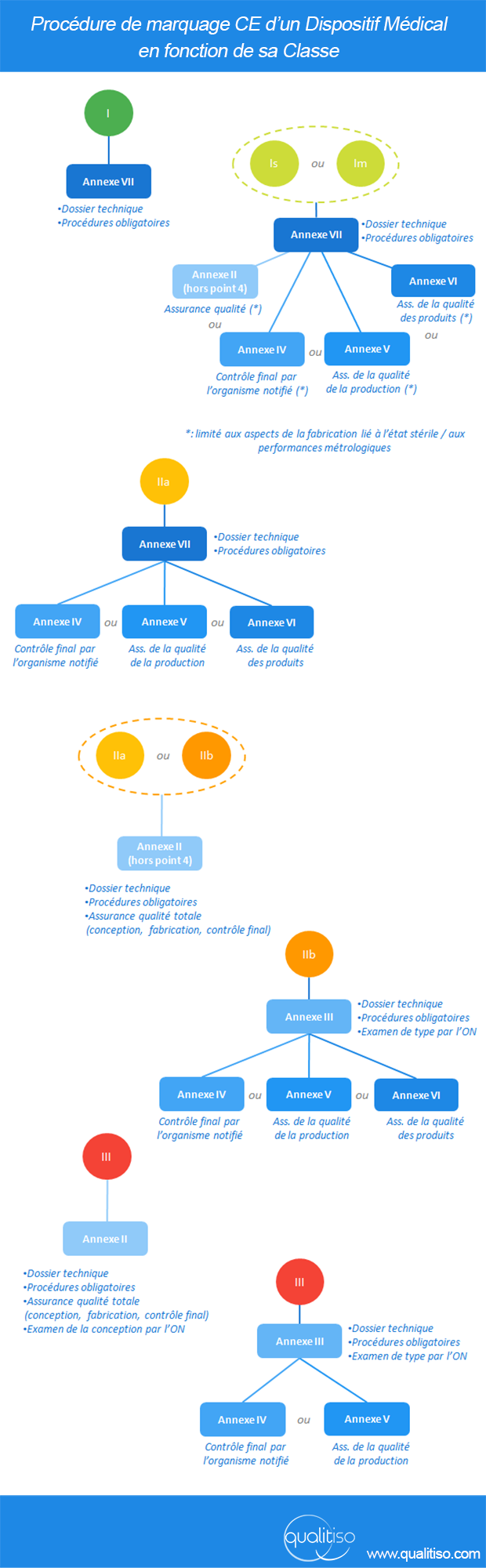
(cliquez pour agrandir)
62 commentaires