Guide pour les porteurs de projet de dispositif médical

Vous portez un projet de dispositif médical ? Vous voulez y voir clair dans ce monde fait de normes et de règlementations ? Vous vous questionnez sur les coûts, les durées et les ressources nécessaires pour mettre sur le marché un DM ?
Ce guide est fait pour vous aider à identifier et à comprendre les exigences qui pèsent sur le secteur.
Bienvenue dans l’aventure du DM !
Attention : le secteur du dispositif médical vis sa révolution ! le règlement (UE) 2017/745 va remplacer la directive 93/42/CEE encore en vigueur, nous sommes à mi-parcours d’une période de transition qui se terminera le 26 mai 2020 et qui chamboule les plannings et les habitudes de tous les acteurs du DM. Ce guide tient compte des deux référentiels.
Quelle règlementation s’applique aux dispositifs médicaux ?

En Europe, les dispositifs médicaux sont définis et encadrés par la directive 93/42/CEE qui sera abrogée en mai 2020 par le Règlement 2017/745.
D’ autres règlementations sont potentiellement applicables à vos projets : c’est le cas si vous utilisez des substances dangereuses, des médicaments, des composants électroniques, si vous fournissez les instructions uniquement en format informatique … Ces réglementations sont générales (applicables à tout produit) mais le gros des exigences vient de la règlementation DM.
Le respect de la règlementation est matérialisé par le marquage CE du dispositif, associé à une déclaration de conformité émise par le fabricant, de durée limitée à 5 ans elle doit être régulièrement renouvelée.
À l’exception des dispositifs à faible risque (de classe I qui sont en auto-certification) l’intervention d’un organisme notifié sera nécessaire pour certifier vos activités et vos produits.
Cas particuliers : les dispositifs sur mesure et les dispositifs destinés aux investigations cliniques ont leur propres exigences.
La règlementation européenne est reprise dans la règlementation française, les règlements sont retranscrits tels quels tandis que les directives peuvent être légèrement remaniées (voir par exemple l’arrêté du 15 mars 2010 qui modifie les exigences essentielles de la directive 93/42/CEE).
Effectuer une veille règlementaire et normative : PRO.GNR
Liste des Règlementations Applicables : OTL.LRA
Mon produit est-il un dispositif médical ?

Le statut de dispositif médical dépend de la finalité revendiquée par le fabriquant, indépendamment des technologies utilisées. Un produit sera DM s’il est, par exemple, destiné à diagnostiquer une maladie où à compenser une blessure. Pour statuer il faut se référer à la définition de DM donnée par la directive et prendre en compte les modifications apportées par le règlement.
Notez que des dispositifs hors finalité médicale sont également concernés par le règlement, ils sont listés dans l’annexe XVI. Ces cas particuliers sont justifiés par leur potentielle dangerosité (ex : des lentilles de contact “fantaisie” ou un dispositif de lipolyse).
Statut de fabricant
Fait original : le fabricant n’est pas obligé de fabriquer le dispositif 🙃
En effet, une entreprise devient fabriquant de dispositif médical lorsqu’il met sur le marché le DM en son nom, peu importe qui l’a conçu ou qui l’a produit.
Cela ouvre la voie aux dispositifs OEM : conçus et fabriqués par un tiers, ils sont mis sur le marché par un fabricant OBL (assez courant avec les petits DM électroniques type oxymètre, tensiomètre ou thermomètre, où la technologie est maitrisée et les prix sont contenus).
Faut-il utiliser la Directive ou le Règlement ?
Tout dépend de votre timing : si vous (ainsi que votre organisme notifié) êtes prêt(s) pour une certification selon la directive alors il est vivement recommandé de le faire : le certificat restera valable après la mise en application du règlement : jusqu’à sa date de fin et le 26 mai 2024 au plus tard. Cette “période de grâce” vous permettra de préparer la transition au règlement en douceur…
Attention, les dispositifs de classe I devront obligatoirement être conformes au règlement dès mai 2020, il est capital de prendre en compte dès à présent les nouvelles exigences.
Quelle est la classe de mon dispositif ?
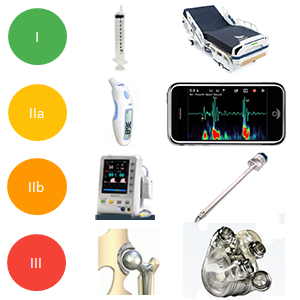
La classe du dispositif est proportionnelle à son niveau de dangerosité potentielle, elle est évaluée par le fabricant (et vérifiée l’ON) selon l’annexe IX de la directive 93/42/CEE ( selon l’annexe VIII du règlement 2017/745) les classes existantes sont, par ordre de dangerosité : I, IIa, IIb et III.
La classe est très critique, car elle détermine les exigences applicables et l’effort pour un fabricant est incomparable entre une classe I et une classe III, il est même parfois nécessaire de revoir les revendications associées au dispositif afin d’éviter une classe trop élevée.
Définir la classe du dispositif selon le règlement DM : OTL.CLS
Définir la classe du dispositif selon la directive DM : OTL.CLS.93.42
Comment obtenir le marquage CE ?
Mettre en œuvre une procédure de marquage CE
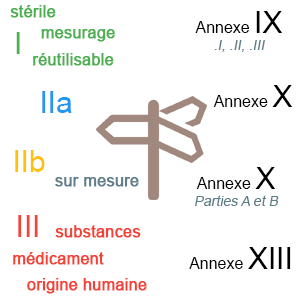
Les procédures d’évaluation de la conformité sont définies dans le règlement (la directive), elles dépendent de la classe du dispositif et de ses caractéristiques, les procédures prévues vont de l’auto-certification à une assurance qualité complète :
| Procédure | Remarque |
|---|---|
| Auto certification | Uniquement les DM de classe I, hors classe Is (stérile), Im (intégrant une fonction de mesure) ou Icr (instruments chirurgicaux réutilisables). Ne nécessite pas l’intervention d’un organisme notifié. |
| Assurance qualité totale | Certification par un organisme notifié du système de management de la qualité complet de l’entreprise |
| Assurance qualité limité à la production ou au contrôle final | Système qualité limité, pour des entreprises mono-produit ou mono-groupe |
| Contrôle final des dispositifs par l’ON | L’Organisme Notifié est sollicité pour chaque libération de lot. Réservé aux productions très ponctuelles |
Un système qualité doit être construit selon la normeISO 13485, une version “renforcée pour le médical” de l’ISO 9001 et dont la version 2016 sera obligatoire dès mars 2019. C’est ce référentiel qui sera utilisé par l’ON pour vous auditer.
- Répondre aux exigences règlementaires prend classiquement de 9 à 18 mois pour une jeune pousse (en parallèle de la construction du SMQ), en fonction de vos disponibilités, de la complexité et de la maturité du projet.
- Votre responsable qualité / règlementaire à temps plein
Travailler avec un Organisme Notifié

L’intervention d’un ON est obligatoire dès la classe IIa (et les classe Is, Im et Icr). Attention aux disponibilités en cette période de transition, les nouveaux acteurs ont parfois des mois d’attente avant d’avoir un organisme prêt à prendre leur dossier… Les organismes notifiés pour la directive sont listés par la commission européenne, le GMED est le seul Français. Les notifications pour le règlement sont en cours et devraient commencer fin 2019 😨
Classiquement, le travail avec l’ON comprend :
- Des échanges autour de l’entreprise et du dispositif
- La formalisation de la prestation
- Un audit de votre système qualité
- Une revue de votre documentation technique
- L’obtention marquage CE 😀
Viennent ensuite les audits de suivi (annuel), les audits de renouvellement (tous les 3 ans pour le SMQ et tous les 5 ans pour le CE du dispositif) et les audits inopinés (au moins un tous les 5 ans).
- Les coûts dépendent de la complexité de votre dossier et surtout de la taille de votre entreprise, comptez au tour de 15’000€ la première année et 5’000€ annuels, pour des projets modestes.
- La durée du travail avec l’ON dépend principalement de votre avancement et dure classiquement de 9 à 12 mois pour une première certification.
Comment construire un système qualité ?

Que vous soyez audité par un organisme notifié ou en auto-certification le règlement prévoit des exigences qui ne peuvent être respectées que par la mise en œuvre d’un Système de Management de la Qualité.
La norme ISO 13485 est la norme de management utilisée en contexte dispositif médical, comme la 9001 elle utilise une approche processus (approche selon vos activités) soutenue par une approche par les risques propres au médical, pour construire un système en proportion des risques induits par vos activités et produits.
Les exigences qualité se regroupent en trois familles :
- Activités de “management” : gestion des ressources humaines, revues de direction, suivi et analyse de vos activités, gestion des non-conformités, …
- Activités de “support” : gestion des infrastructures, des équipements de mesure, des documents, des achats, des instruments de contrôle et mesure, …
- Activités “opérationnelles” : conception et développement, production, prestations de service, surveillance après commercialisation,…
Ce travail impacte tous les services et tous les acteurs de l’entreprise, il est orchestré par le responsable qualité.
- de 5k€ à 20k€ pour l’accompagnement et les formations
- de 6 à 18 mois en fonction de l’accompagnement et de votre autonomie; le SMQ est construit en parallèle de la documentation technique
- votre responsable qualité< à temps plein/li>
Templates pour mettre en œuvre l’ISO 13485
Quelles exigences s’appliquent à mon dispositif ?
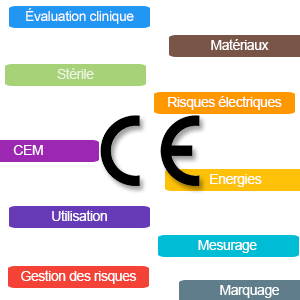
Vos dispositifs doivent respecter les exigences générales de sécurité et de performance, elles sont définies dans l’annexe I du règlement (l’équivalent des exigences essentielles de la directive), il vous faudra évaluer et justifier leur (non)applicabilité. Ces réponses seront la “porte d’entrée” pour quiconque examine votre dossier, en premier lieu les auditeurs.
Les exigences gravitent autour de deux sujets fondamentaux :
- Prouver la sécurité du dispositif : essais de sécurité électrique, biocompatibilité, évaluation biologique, validation des activités de stérilisation …
- Prouver les performances du dispositif : dossier d’évaluation clinique avec des données de la littérature et/ou des données issues d’investigations cliniques (une grosse tendance du nouveau règlement)
Ces exigences sont respectées en mettant en œuvre des normes, des spécifications communes ou d’autres référentiels reconnus par la règlementation et les organismes notifiés.
- Les coûts sont ceux associés à la mise en œuvre des normes :temps humain, coûts des essais laboratoire, organisation des validations …
- La prise en compte des normes se fait durant toute la création de la documentation technique, puis durant toute la vie du dispositif
- Un responsable qualité/règlementaire à temps plein, les chefs de services sont sollicités pour mobiliser leurs services pour la prise en compte des normes (ex : le bureau d’étude)
Quelles sont les normes à respecter ?

Des normes harmonisées (actuellement à la directive, prochainement au règlement) sont identifiées par la commission européenne, une liste est tenue à jour en fonction des évolutions normatives, leur mise en œuvre fait preuve de présomption de conformité, un recours quasi-obligatoire, surtout si un organisme notifié est impliqué.
Les autres normes (non harmonisées) sont à appliquer si aucun référentiel ne permet de répondre aux exigences, d’autant plus si la norme existe en version EN (elle est alors reconnue pour répondre au contexte règlementaire européen).
Les spécifications communes seront publiées par la commission européenne, en vue d’apporter des réponses sur des points non couverts par les normes.
- – Achat des normes : en fonction du nombre de normes applicables, des versions achetées et de la boutique choisie cela va de quelques centaines d’euros à plusieurs milliers, attention au cycle de vie des normes qui sont revues tous les 5 ans.
– Formations : comptez une journée par sujet (SMQ, règlementation, logiciel, gestion des risques, évaluation clinique,…) - La veille peut nécessiter plusieurs jours par an, surtout lorsqu’il s’agit d’approfondir de nouveaux sujets
Garantir les performances
En plus des réponses aux normes générales vos dispositifs seront probablement concernés par des normes spécifiques, comme l’IEC 62304 pour tous les logiciel (de) dispositif médical ou l’IEC 60601-1 pour les dispositifs électromédicaux, voire des normes propres à des types de DM, comme pour les implants de hanche, les raccords, le matériel ophtalmique…
Les exigences sont prises en compte lors des activités de R&D, leur respect est vérifié par des tests en interne et en ayant recours à des essais en laboratoire.
Les laboratoires sont choisis en fonction de leurs accréditations pour les référentiels de test (certificat COFRAC par exemple).
- de 5’000€ à plusieurs k€ en fonction des essais à réaliser en laboratoire
- les essais prennent de quelques jours à quelques mois en cas de non-conformité à corriger
- de 2 à 12 hommes/mois pour la préparation et le suivi
Templates applicables aux logiciels (embarqué ou autonomes) de dispositifs médicaux
Templates pour les dispositifs électro-médicaux
Garantir un rapport bénéfice/risque favorable
L’établissement et le maintient d’un rapport bénéfice/risque favorable est l’exigence au cœur de la règlementation.
Cela passe par des activités de gestion des risques qui seront identifiés et maitrisés, et par la gestion des bénéfices patients, afin de mener à bien l’évaluation du rapport bénéfice/risque du dispositif.
La norme ISO 14971 est utilisée pour les activités de gestion des risques et est appelée par la quasi-totalité des normes médicales.
- de 1 à 6 homme/mois pour une première version du dossier, puis une douzaine de jours par an pour le faire vivre, voire plus en cas de nouveauté, d’incident…
Templates pour la gestion des risques
Gérer les erreurs d’utilisation
Les risques liés aux erreurs d’utilisations potentielles des utilisateurs sont traités selon des activités d’ingénierie de l’aptitude à l’utilisation, selon la norme IEC 62366-1.
Les interactions de l’utilisateur avec le dispositif sont analysées, l’aptitude à l’utilisation est évaluée au fil du développement et en fin de R&D lors des activités de validation, en “conditions réelles” (mais maitrisées).
- les coûts dépendent de vos besoins externes pour planifier et superviser les évaluations finales (living lab, recrutement d’utilisateur, experts, mise à disposition de locaux et de matériel, ..) : comptez de 2 à 10k€
- de 1 à 6 homme/mois
Templates pour l’ingénierie de l’aptitude à l’utilisation
Fournir les bonnes informations à l’utilisateur

En plus de présenter et d’expliquer votre dispositif les IFU (instructions et étiquetage) garantissent une utilisation sûre et efficace de votre DM.
Des mentions (public visé, contre-indications, “que faire en cas de défaillance”,…) et des marquages (plage de température d’utilisation, type d’alimentation électrique, date de fabrication…) sont normalisés et obligatoires.
Ces éléments seront contrôlés par les laboratoires et l’organisme notifié.
Définir les exigences sur l’étiquetage : OTL.ETQ
Définir les exigences pour les instructions fournies à l’utilisateur : OTL.INS

Faut-il réaliser une étude clinique ?
La sécurité et les bénéfices patients doivent être démontrés, les données utilisées sont de nature pré-clinique (les essais en interne et en laboratoire) et clinique.
Les données cliniques sont générées via les activités d’évaluation clinique.
S’il est possible de trouver les informations nécessaires dans la littérature , en s’appuyant sur les données acquises sur une autre version du dispositif, ou en faisant une équivalence avec un autre DM, il faudra néanmoins souvent recourir à une investigation cliniques : des essais en conditions réelles, sur des patients, encadrés par votre autorité compétente : l’ANSM en France.
Dans tous les cas, le dossier d’évaluation clinique est rédigé selon l’état de l’art, notamment le guide MEDDEV 2.7/1 rev4.
Les données cliniques sont ensuite mises à jour durant toute la vie des dispositifs, selon les activités de surveillance clinique après commercialisation, pour affiner l’analyse du bénéfice/risque et maintenir la conformité du dispositif.
- plusieurs dizaines de milliers d’euros pour une investigation clinique (essais sur patient) et des frais associés (assurance, investigateur…)
- de quelques mois pour une évaluation documentaire à plusieurs semestres pour une investigation clinique
Templates pour l’évaluation clinique et le suivi clinique après commercialisation des dispositifs médicaux
Comment obtenir le remboursement de mon dispositif ?
Le remboursement d’un DM n’est pas systématique, il est basé sur une évaluation de son intérêt médico-économique, plusieurs options se présentent pour prétendre au remboursement et une inscription à la LPPR.
Prise en charge dans le cadre d’un usage individuel :
- Inscription générique : le dispositif doit répondre aux critères bien définis pour entrer dans la ligne de remboursement
- Inscription en nom de marque : le remboursement est réservé à une unique référence de dispositif, le fabricant a porté le dossier et verrouille ainsi le remboursement
- plusieurs k€,
- de 12 à 24 mois
- 1 homme/an
- Fabricant : vous, qui mettez sur le marché un dispositif médical en votre nom
- Organisme notifié : l’organisme notifié vis-à-vis de la règlementation pour venir vous casser les pieds
- Auditeur : le casse-pieds de l’ON
- Autorité compétente : l’ANSM en France
- Sous-traitant critique : une entreprise qui impacte la sécurité ou les performances de votre dispositif (ex : qui prend en charge la conception, la fabrication, la maintenance…)
- Consultant : une personne payée trop cher pour vous aider à résoudre des problèmes que vous n’aviez pas avant de lire cet article
- Formateur : consultant en mode “maitre d’école”, grand fan de votre OPCA
- Distributeur : organisme qui met à disposition le DM, à l’utilisateur ou à un autre distributeur
- Importateur : organisme qui met sur le marché UE un DM dont le fabricant est hors UE
- Mandataire : le représentant légal d’un fabricant hors UE (obligatoire)
Prise en charge dans le cadre d’un acte : les contraintes sont alors beaucoup plus importantes.
À noter : un forfait innovation est proposé pour accélérer la mise sur le marché des dispositifs médicaux innovants, l’évaluation est alors affinée après mise sur le marché, pour confirmer l’intérêt médico-économique.
Quelles sont les autres obligations d’un fabricant ?
Les dispositifs mis sur le marché sont communiqués à l’ANSM, en attendant la base électronique européenne Eudamed prévue par le règlement.
Les volumes de ventes sont également notifiés à l’ANSM chaque année.
Une assurance est requise contre les produits défaillants (directive 85/374/CEE), vous pouvez également choisir un contrat tenant compte des problématiques du secteur (ex : assurance contre la perte de marquage CE).
Des communications et des échanges d’informations sont prévus avec les organismes notifiés et les autorités nationales, via Eudamed.
Enfin : la surveillance après commercialisation est à mener continuellement, pour vérifier les performances et la sécurité de vos produits, et mener des actions d’amélioration au besoin.
En cas d’incident les activités de vigilance assurent une analyse des non-conformités, qui déclenchent des actions correctives à mener sans délais indus.
Surveillance après commercialisation : PRO.SAC
Vigilance : PRO.VGL
Gestion des communications règlementaires : PRO.COM
Liste de documents utiles : OTL.LDU
Annexes
Le parcours du DM

28 commentaires