Format IMDRF “Table des matières” pour la soumission des dossiers règlementaires des dispositifs médicaux
L’IMDRF a publié en le 20 mars 2019 un guide pour la soumission des dossiers techniques des dispositifs médicaux selon le format IMDRF.
Ce format (spécifiant des dossiers, sous dossiers et documents attendu) se veut international (US FDA, Santé Canada, TGA, ANVISA, Chine, Europe, Japon, Russie, Singapour et Corée du Sud) il permet de tenir compte des dossiers exigés par les différentes règlementations. C’est le successeur du format STED porté en son temps par le GHTF.
L’utilisation d’un tel format permet de rationaliser vos dossiers de soumission selon les différentes règlementations, chaque autorité (FDA, Santé Canada…) propose ou proposera une matrice de correspondance entre ses propres exigences et le format défini par l’IMRDF.
Ce format est particulièrement très utile si vous répondez à plusieurs règlementation et/ou si vous êtes en mal d’inspiration pour organiser votre documentation technique et plus généralement les données de sortie de vos projets. C’est un complément précieux de la documentation technique exigée par le règlement (UE) 2017/745 définie de façon assez peu pratique dans l’annexe II et l’annexe III du RDM.
Présentation des documents utiles
Le projet est géré par l’IMDRF (voir la page des documents de l’IMDRF) mais implique les principales autorités règlementaires. Les documents et modèles associés sont listés ci-dessous :
- IMDRF : Templates relatifs au modèle de dossier IMRDF (20/03/2019)
- Santé Canada : Ébauches des Lignes directrices de Santé Canada pour les demandes d’homologation d’instruments médicaux fondées sur la table des matières de l’IMDRF (28/02/2019)
- Santé Canada : Matrice de correspondance (28/02/2019)
- MIDRF : Guide pour les templates (24/01/2019)
- FDA : Matrice de correspondance (08/08/2018)
- TGA : Matrice de correspondance (27/03/2018)
- IMDRF : Détails sur le modèle de dossier de soumission (27/03/2018)
Les matrices de correspondance sont extrêmement importantes : elles définissent l’applicabilité des exigences selon la règlementation appliquée et précise le contenu attendu dans les différents dossiers. Pour l’instant la FDA, le TGA et Santé Canada sont les autorités les plus actives pour le projet de l’IMDRF.
Le modèles IMDRF sur qualitiso

Retrouvez la table des matières IMDRF dans l’outil OTL.IMDRF.TOC, il comporte les remarques du guide de l’IMDRF, prend en compte la matrice de correspondance de Santé Canada et fait le lien avec les exigences du règlement DM en matière de documentation technique (générale et relative à la surveillance après commercialisation).
La suite de l’article reprend l’organisation du modèle IMDRF et décrit le contenu attendu pour chaque rubrique.
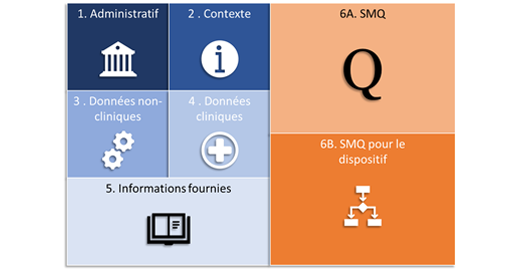
1. Renseignements administratifs régionaux
Ce dossier est le plus spécifique à chaque réglementation : il contient les différents échanges, formulaires, lettres et tout document administratif exigé par les autorités.
C’est également dans ce dossier que le fabricant identifie les certifications et conformités qu’il revendique, en joignant les certificats associés, ou encore le certificat de libre vente délivré par l’autorité compétente
2. Contexte
On retrouve les informations sur le dispositif : description (du DM et de son emballage), historique des versions précédentes (dont les chiffres de vente et d’incidents), principes de fonctionnement et dispositifs équivalents.
L’utilisation prévue du dispositif est également définie : indication, environnement, contre-indications…
3. Données non-cliniques
Toutes les données utilisées pour démontrer la conformité du dispositif, durant vos vérifications et validations, qui ne sont pas de nature clinique.
Cette partie inclut la gestion des risques et les normes applicables au dispositif.
Liste des études mises en oeuvre
Les données techniques (les études) sont présentés de manière systématique :
- Description, identification, dates
- Résumé
- Rapport complet
- Données statistiques (pour l’US FDA)
Les données couvrent les champs suivants :
- Caractérisation physique et mécanique
- Caractérisation chimique et matérielle
- Systèmes électriques : sécurité, protection mécanique et environnementale et compatibilité électromagnétique
- Radioprotection
- Évaluation de la biocompatibilité et de la toxicologie
- Pyrogénicité non liée au matériau
- Sécurité du matériel biologique d’origine humaine et animale
- Test sur les animaux
- Utilisabilité et facteurs humains
liste à laquelle il faudra ajouter une section “fonction de mesurage”, lorsqu’utile.
Les logiciels ainsi que la validation de la stérilisation profitent d’une organisation plus détaillée, en accord avec les données de sortie exigées par les normes applicables.
Période d’expiration et validation de l’emballage
Cette partie permet de renseigner les tests de stabilité du produit que vous auriez réalisés, ainsi que les résultats de la validation des emballages.
4. Données cliniques
Ce dossier accueille le résultat de votre évaluation clinique :
- Rapport d’évaluation clinique
- Investigations cliniques
- Données de suivi après commercialisation
- Données de la littérature
5. Étiquetage et matériel promotionnel
Cette partie du dossier contient toutes les informations fournies aux utilisateurs, sous toutes leurs formes :
- Instructions d’utilisation
- Étiquettes
- Informations sous format électronique
- Informations à destination des professionnels de santé
- Informations à destination du patient
- Manuel technique
- Autocollant / carte / fiche d’implant
- Brochures commerciales
- Autre étiquetage
- Autre matériel promotionnel
6A. Système de management de la qualité
Il est essentiellement question d’identifier les procédures de l’entreprise en rapport avec les chapitres 4 à 8 de la norme lSO 13485:2016, notamment :
- Manuel qualité
- Procédure de gestion des documents
- Procédure achats
- Procédure de gestion des équipements
- …
6B. Documentation qualité associée au dispositif
Cette dernière section est dédiée aux documents qualité dédiés à la réalisation du produit (les données de sortie de la mise en œuvre de votre SMQ) :
- Enregistrements liés à la R&D
- Spécification et contrôle des achats pour le dispositif
- Process de fabrication et contrôles réalisés
- Indicateurs et analyse, surveillance du produit
Conclusion
Cette structure de dossier de soumission règlementaire reprend les exigences communes des règlementations sur les DM tout en offrant des moyens de réponse aux exigences particulières.
C’est un excellent moyen de présenter votre documentation en vue de multiples procédures règlementaires, qui vous simplifie la gestion de multiples dossiers et rassurera vos auditeurs quant à la qualité de l’organisation de votre information.
L’IMDRF a de grandes ambitions pour son modèle de dossier de soumission, qui pourrait être à la base de futures soumissions électroniques que visent les différentes autorités.
4 commentaires