
Nouveau site internet pour le GMED
Lifting du site internet du GMED, qui gagne en contenu et en clarté. Parmi les rubriques utiles : Les accréditations du GMED; Un espace de.
Lifting du site internet du GMED, qui gagne en contenu et en clarté. Parmi les rubriques utiles : Les accréditations du GMED; Un espace de.
Tendance à la baisse depuis 2010 pour certains indicateurs majeurs en France, relatifs à la santé, l’éducation, la sécurité ou l’économie.

Restez informé des nouvelles publication via les alertes mail

Tous anti-covid : statut règlementaire et efficacité

Guide et informations relatives à l’enregistrement des opérateurs économique sur la base Eudamed.

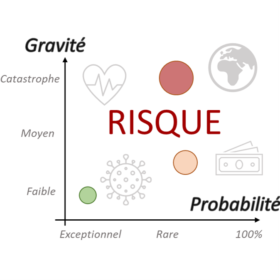
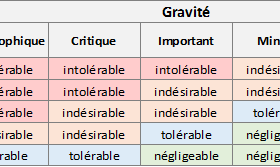
Forme des risques et acceptabilité
Guide HAS pour le dépôt de dossier de remboursement
Guide relatif à la vigilance pour les fabricants de pompes à insuline
IEC 62366-1/A1 : résumé des changements, comparatif des prix

Rapport 2020 de l’observatoire étudiant des violences sexuelles.

Nouvelle montre Apple, nouvelle fonctionnalité de santé, nouvelle arnaque médicale
5 indicateurs pour mesurer le monde, sans recourir au PIB.

Le monde selon ses principaux indicateurs, hors PIB : Santé, Démographie, Pauvreté, Progrès et Pollution.
Explication de la méthode utilisée dans le cadre de l’analyse des risques du monde.
Réglementations applicables, normes et recommandations pour les fabricants d’intelligences artificielles médicales.
Impact du PLFSS 2021 sur le remboursement des dispositifs médicaux

FAQ relative à la crise du coronavirus et à sa gestion en France.

Amendement 1 de septembre 2020 pour la norme IEC 60601-1-2 : perturbations électromagnétiques – Exigences et essais.
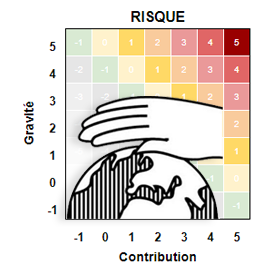
Techniques et formules utilisées sur calculer les risques. Cette approche est tirée des normes ISO 14971 et XP S99-223, l’analyse des risques est universelle : elle.
Notification de 3EC International a.s pour le règlement (UE) 2017/745
Quel modèle économique pour la transition écologique ? Gaël Giraud est l’invité d’Etienne Klein.

Revue des différentes réponses des medtechs à la crise du coronavirus, par Mathilde Béal.
Le projet de respirateur 100% Marocain, lancé en mai dernier en début de crise covid, n’est toujours pas disponible, le blocage est d’ordre réglementaire. Des.
Version 2020 du référentiel HAS pour les LAP en enquête publique
Rappel de la MHRA sur la conformité des dispositifs de classe I
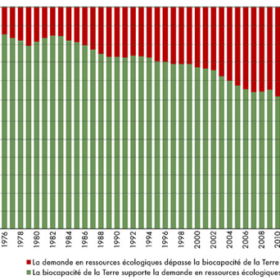
Recul de la date du dépassement en 2020

Avis de la HAS sur le remboursement des masques chirurgicaux Yingmed dans le contexte de la covid-19
Modification des systèmes de délivrance d’insuline : attention danger
Résumé du guide ISO/TR 20416 relatif à la surveillance après commercialisation des dispositifs médicaux
Règlement d’exécution (UE) 2020/1207 : spécifications communes pour le retraitement des dispositifs à usage unique.
Notification de DQS Medizinprodukte GmbH pour le règlement (UE) 2017/745

Disponibilité et capacité des organismes notifiés à effectuer des évaluations de conformité pour les dispositifs médicaux liés au COVID-19

Bilan 20019 pour l’industrie du dispositif médical
Grille d’analyse des évaluation cliniques des dispositifs médicaux
Révision 2020 de la norme ISO 14155 : Investigation clinique des dispositifs médicaux pour sujets humains — Bonne pratique clinique
Guides et check-listes pour la restérilisation et le retraitement des dispositifs médicaux.

Guide autour de la notion de «Risque» : définitions utiles, les grands types de risques et les méthodes pour la gestion des risques. Le champ est multiple : risques sanitaires, risques en entreprise, risques socio-économique, risques climatiques …

Obligations autour des masques de protection grand public et étude du statut règlementaire
Informations relatives aux masques en contexte coronavirus : masques FFP, masques chirugicaux / médicaux, masques artisanaux, masques barrières : utilisation, spécifications, performances, obligations.
Nouveau site de l’UE pour les dispositifs médicaux
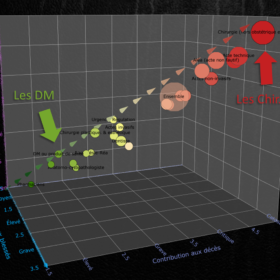
Bilan des risques médicaux en France : causes des incidents, spécialités concernées, conséquences pour les patients et conséquences financières
Enquêtes en cours sur les normes utiles dans le médical
Rapport 2019 sur les incidents de sécurité des systèmes d’information pour le secteur santé

Revue de la littérature de la TGA autour des risques associés aux logiciels médicaux.

Notification du GMED pour le règlement dispositifs médicaux

Exigences et aspects pratiques concernant l’enregistrement des dispositifs médicaux à l’international.
Publication du guide ISO/TR 24971, pour la norme ISO 14971 relative à la gestion des risques des dispositifs médicaux.
Règlements DM et DM-DIV : rejet du mandat de normalisation par le CEN CENELEC
Guide du MDCG pour la consultation d’une autorité dans le cadre de l’évaluation d’un dispositif médical combiné

Retour d’expérience sur la qualification des logiciels utilisés par l’entreprise : exemple avec un ERP
Mesures pour faciliter le renouvellement des désignations des organismes notifiés pendant la crise covid.
Principes pour des prestations de santé garantissant la dignité des personnes âgées, norme EN 17500

Risques économiques en France associées à la crise du coronavirus : impact par secteur d’activité, chômage, dette, CAC. Évaluation du rapport Bénéfice patient / Risque économique.

Réglementation environnementale pour les entreprises et leurs produits : substances dangereuses, déchets, emballages, piles, appareils électriques. Utilisation de l’éco-conception dans une optique de développement durable et Responsabilité élargie des producteurs.

[Guide] Réduire les risques pour l’environnement grâce à la bonne conception de vos dispositifs.
Mandat de normalisation CEN CENELEC pour les normes harmonisées au règlement (UE) 2017/745
Formulaire MIR pour la déclaration d’incidents et FAQ associée
Procédure d’autorisation de mise sur le marché de dispositifs médicaux par dérogations aux règles de marquage CE.

Stratégies pour le règlement (UE) 2017/745, à destination des fabricants
Analyse et maitrise des risques environnementaux associés aux produits et services.

Présentation des normes pour le management environnemental : ISO 14001, ISO 14006, IEC 62430… points importants, remarques et liens utiles
Liens pour identifier les produits frauduleux ou vérifier les certificats CE des dispositifs médicaux et les certificats ISO 13485 des fabricants.
Informations sur la disponibilité des modules Eduamed
Notification de Intertek Medical Notified Body AB pour le règlement (UE) 2017/745

Déroulement d’un audit à distance en période de confinement, par Benoit Daclin.

Archives audio des épisodes de la saison 2019-2020 du podcast Qualitiso.
Rapport team-NB 2019 sur les ressources et activités des organismes notifiés.
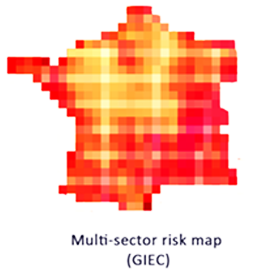
Analyse des risques associés au réchauffement climatique, analyse bénéfice/risque des investissements financiers nécessaires pour contenir à +2°C.
Analyse de l’historique des risques de terrorisme en France. Données sur les attentats islamistes (les mieux documentés). Carte des risques Évolution temporelle Corrélation du niveau.
Analyse des risques : Accidents de la route en France

Compilation d’informations utiles pour concevoir et fabriquer un respirateur pour patients en contexte covid19.

Loi sur l’économie circulaire et exigences écologiques en contexte dispositifs médicaux.

Analyse des risques de violences en France

Préoccupations des Français, risques associés, historique et corrélations
MDC MEDICAL DEVICE CERTIFICATION GMBH, (ON Allemand N°0483) vient d’être notifié pour le règlement 2017/745.

Les dates clés pour la mise en oeuvre du règlement (UE) 2017/745
Preuves cliniques nécessaires pour les dispositifs CE selon la directive 93/42/CEE, dans le cadre de la transition au règlement (UE) 2017/745.
Le règlement (UE) 2017/745 est décalé d’un an.
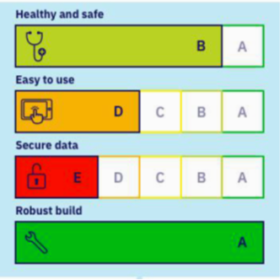
Logiciels de santé : classification en niveaux A, B , C et D selon la HAS

Analyse des Risques globaux mondiaux

Analyse des causes de décès en France : nature, répartition par sexe et par âge
Le MDCG a publié un guide pour l’article 54.2.b du règlement DM relatif à la procédure de consultation dans le cadre de l’évaluation clinique des dispositifs de classe III ou IIb destiné à administrer ou retirer un médicament.

Informations pour mise sur le marché de dispositifs médicaux en contexte coronavirus

Covid-19 / Lunettes de protection – informations techniques, normatives, réglementaires
Règlement (UE) 2017/745 vers un décalage d’un an
Nouvelles normes harmonisées à la directive 93/42/CEE
CE Certiso Orvos, (ON Hongrois N°2409) est notifié pour le règlement (UE) 2017/745.

Mise à jour du suivi de l’avancement des évaluations des ON, pour leur notification au règlement (UE) 2017/745
Principes clés pour l’évaluation clinique des logiciels (de) dispositifs médicaux, selon le guide MDCG 2020-1.

Mesures transitoires pour les dispositifs médicaux de classe I, avant la mise en œuvre du règlement (UE) 2017/745.
Guide pour l’analyse de l’aspect “significatif” des changements apportés à un dispositif médical
Mise à jour du guide du MDCG relatif à l’IUD-ID de base et ses modifications.
Plan de mise en œuvre/préparation du nouveau règlement sur les dispositifs médicaux 2017/745. Ce document présente un plan conjoint de la Commission et des États membres, comprenant des actions prioritaires et d’urgence pour mettre en place un système opérationnel d’ici mai 2020.
Exigences et modèles pour les cartes d’implant des dispositifs médicaux : carte et livret associé
Mesures du GMED pour la gestion de la crise liée au coronavirus du, focus sur le document IAF ID3.
Questions/Réponses à destination des fabricants de dispositifs médicaux de classe I, prise en compte du règlement (UE) 2017/745 et de la directive 93/42/CEE.
compte rendu de la réunion du 6 février 2020 Comité d’interface ANSM / Organisations professionnelles Représentatives des industries des DM et des DMDIV : Groupe de Travail Eudamed
Suites de l’affaire PIP avec les conclusions de la Cour fédérale de justice allemande
Nouveaux symboles de la norme ISO 15223-1:2020
Les normes ISO 10993-2 et ISO 10993-10 sont en enquête publique
Proposition de la TGA pour un nouveau régime réglementaire pour les dispositifs médicaux personnalisés, y compris les dispositifs imprimés en 3D.
FAQ du MDCG relative aux DM sur mesure et aux DM adaptables
Résumé de la note de la TGA relative au Brexit. Les informations sont largement valables pour l’Europ

Résumé de la norme expérimentae française XP S99-223 : Dispositifs Médicaux – gestion du rapport Bénéfice/Risque
La HAS a mis à jour sa procédure d’élaboration des avis du Collège de la HAS en vue de la prise en charge dérogatoire d’un DM/DMDIV/acte innovant.
L’ANSM a publié le compte rendu du Groupe de travail « Règlements DM/DMDIV », associé au Comité d’interface ANSM / Représentants des industries des DM et des DMDIV, qui s’est réuni le 10 septembre 2019.
La première lecture du projet de loi sur les médicaments et les DM a été faite à la Chambre des Communes afin de présenter le nouveau cadre que souhaite instaurer le gouvernement britannique en matière de santé.
L’article L5212-1-1 du CSP suit les ambitions de la réfore du financement de système de santé, en encadrant la remise en bon état d’usage de certains DM à usage individuel.

Une série de chiffres et de graphiques sur les dispositifs médicaux en France: entreprises, coût, certification, emploi,…
Norme IEC 63120 relative au reconditionnement et la réutilisation des dispositifs (électro)médicaux.
ISO 10993-X pour la biocompatibilité des dispositifs médicaux
Impact de la la loi n° 2020-105 fixant les exigences en matière de lutte contre le gaspillage et d’économie circulaire.
L’ANSM a mis à jour le formulaire de déclaration de ventes annuelles des DM et de DMDIV réalisées en 2019.
La présentation et la notice explicative qui accompagnent le formulaire ont été mises à jour.
Cette déclaration est à effectuer avant le 31 mars 2020 à declarationventes.DM-DMDIV@ansm.sante.fr.
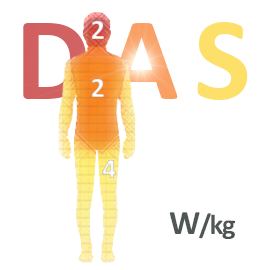
Obligation d’affichage du DAS sur les équipements
Le National Standards Authority of Ireland vient d’être notifié pour le règlement (UE) 2017/745, les (nombreuses) exclusions sont listées ci-dessous
Programme de travail de l’UE dans le domaine de la santé, pour 2020
Avis préliminaire sur la protection des données pour la recherche scientifique, de l’EDPS.
Mise à jour du draft du guide MDCG relatif au PSUR

Notification de DNV GL Presafe AS pour le règlement (UE) 2017/745
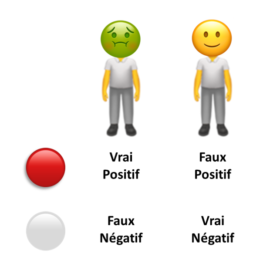
Probabilités appliquées au diagnostic médical, cas des intelligences artificielles
Suppression de la taxe sur le chiffre d’affaires des DM et nouvelle taxe pour les DM inscrits sur la liste en sus : la clause de sauvegarde.
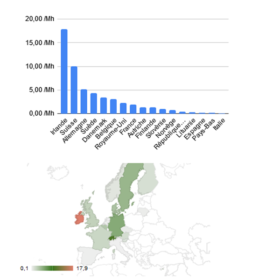
Chiffres et évolutions des NCARS en Europe, données brutes et analyses
La TGA a mis en ligne une page expliquant les principales modifications de la règlementation en matière de produits de santé, dont les DM, à l’occasion du premier ensemble de modifications
Formulaire en ligne des déclarations de vente 2019 de dispositifs médicaux
Un appel à experts pour communications sur le RDM est relié par le team NB
Un document de l’UE fournit des précisions sur la nomenclature CND
Un document de l’UE apporte des précisions sur la nomenclature EMDN des dispositifs médicaux

Résumé du guide MDCG relatif à la cybersécurité des dispositifs médicaux
Le Décret n° 2019-1530 du 30 décembre 2019 relatif à la transparence des liens d’intérêts modifie l’article R1453-3 du CSP
Publication du Décret n° 2019-1452 du 24 décembre 2019 modifiant les procédures applicables au titre de la prise en charge forfaitaire prévue à l’article L. 165-1-1 du code de la sécurité sociale (forfait innovation)

Une communication a été publiée dans Radiology, elle précise une méthode pour évaluer les études utilisant une IA en contexte radiologie

Mémo sur les alarmes des dispositifs médicaux, à destination des utilisateurs et des concepteurs. Selon la norme IEC 60601-1-8 et ses amendements.

L’organisme MEDCERT ZERTIFIZIERUNGS- UND PRÜFUNGSGESELLSCHAFT FÜR DIE MEDIZIN GMBH vient d’être notifié pour le règlement dispositifs médicaux.

Le MDCG a mis à jour sa feuille de route, les changements sont identifiés ci-dessous

Le CCMO a publié une liste des principaux changements causés par le RDM en matière d’investigation clinique, il sont résumés ci-dessous.

Le rolling plan proposé par l’UE a été mis à jour, il fait le point sur les étapes achevées et à venir pour la mise en application des règlements DM et DM-DIV.

Le ministère de la santé a diffusé une fiche d’information relative à la modification des modalités de facturation des dispositifs médicaux implantables inscrits sur la liste en sus.
La norme ISO 11737-2:2019 vient d’être révisée, elle est relative aux contrôles de stérilité pratiqués au moment de la définition, de la validation et de la maintenance d’un procédé de stérilisation.
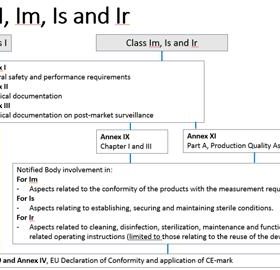
Guide du MDCG pour les fabricants de dispositifs médicaux de classe I, Is, Im et Icr.
Nouvelles publications pour 3 normes relatives à l’évaluation biologique :
ISO 10993-7:2008/Amd 1:2019 – Évaluation biologique des dispositifs médicaux — Partie 7: Résidus de stérilisation à l’oxyde d’éthylène — Amendement 1
ISO 10993-15:2019 – Évaluation biologique des dispositifs médicaux — Partie 15: Identification et quantification des produits de dégradation issus des métaux et alliages
ISO 10993-9:2019 – Évaluation biologique des dispositifs médicaux — Partie 9: Cadre pour l’identification et la quantification des produits potentiels de dégradation
L’arrêté du 10 décembre 2019 remplace, à compter du 1er janvier 2020, les codes LPP attachés à certaines descriptions génériques par les codes d’identification individuelle.
Le parlement adopte le 2nd rectificatif au règlement (EU) 2017/745 relatif aux dispositifs médicaux
Publication de l’ISO 14971:2019
DEKRA Certification B.V. vient d’être notifié pour le règlement (UE) 2017/745, les exclusions sont listées ci-dessous
Guide du MDCG définissant les codes MDA, MDN, MDT et MDS ainsi que leur utilité
Guide du MDCG relatif à l’échantillonnage des dispositifs médicaux de classe IIa et IIb, pour l’évaluation par les ON

Résumé des points relatifs aux DM, lors de la session publique de la commission européenne du 9 décembre dernier, sur le thème de la Santé.
“Lignes directrices à l’intention des fabricants” pour satisfaire aux exigences de l’article 78 de la directive 2013/59 « fixant les normes de base relatives à la protection sanitaire contre les dangers résultant de l’exposition aux rayonnements ionisants »
De nouveaux documents sont disponibles sur le site de la commission, ils donnent des informations très techniques sur l’IUD : le format des codes et les éléments que vous pourrez utiliser pour l’étiquetage des dispositifs.
Dekra NL serrait notifié pour le RDM d’ici quelques jours.
Le CAMD a publié un courrier dans lequel il s’inquiète de la situation engendrée par le décalage de deux ans d’Eudamed : résumé des points durs et des propositions.
Présentation du nouveau format pour la liste des normes harmonisées UE
Livre blanc du BSI relatif aux règlement DM-DIV et à ses conséquences
Le draft du second correctif pour le règlement (UE) 2017/745 est disponible. Attention : il doit encore être approuvé. Modifications Rendez-vous à la page 49.
Norme IEC 80001-1 pour la gestion des risques des dispositifs médicaux connectés à des IT
Projet de notice de la HAS pour l’évaluation de dispositifs médicaux ayant recours à une intelligence artificielle
La HAS vient d’actualiser son guide intra-GHS destiné aux dispositifs médicaux à usage individuel qui sont utilisés en établissement de santé et qui sont financés au travers des prestations hospitalières par une enveloppe globale au titre des Groupes Homogènes de Séjour (intra-GHS).
Revue des changements du guide IMRDF pour les investigations cliniques sur les dispositifs médicaux
L’IMDRF a mis à jour son guide lié à l’évaluation : liste des modifications
Mise à jour du guide IMDRF lié à l’évaluation clinique et ses définitions et concepts clé, ayant pour base la version de 2007 (GHTF/SG5/N1R8:2007) encore sous format GHTF
Le CEPS a développé la plateforme numérique MedimedDM pour faciliter le dépôt des dossiers relatifs aux DM et prestations en vue de leur inscription sur.
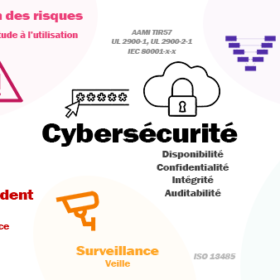
Cybersécurité des dispositifs médicaux : état de l’art, guides et normes appliquables, processus de gestion de la cybersécurité.
Le ministère de la santé a publié le tableau Excel indiquant les nouveaux codes individuels LPP attribués à chaque fabricant qu’il a lui-même déclaré sur.
BSI Group The Netherlands B.V. vient d’être notifié pour le règlement RDM

Les exigences de la présente norme fournissent un cadre pour l’évaluation de l’acceptabilité du rapport bénéfice/risque des dispositifs médicaux. La présente norme a été développée spécifiquement pour les fabricants de dispositifs médicaux.
DARE!! Services B.V. vient d’être notifié pour le règlement RDM.
La HAS met à jour le Guide pour le dépôt d’un dossier auprès de la CNEDiMTS (octobre 2019), ainsi que la matrice du dossier. Pour.
Le ministère de la santé a mis à jour la page internet dédiée à l’identification individuelle pour une inscription en ligne générique des dispositifs médicaux..
Fiche pratique HAS : télémédecine, applis et objets connectés de santé, certification des logiciels métiers (LAP et LAD)
Eudamed décalé de 2 ans.
Découplage de la norme ISO 14971:2019 des accords de Vienne

Revue, résumé et analyse du guide du MDCG pour les logiciels dispositifs médicaux : qualitfication et classification des softs
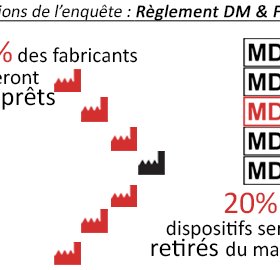
Conclusions de l’enquête Règlement (UE) 2017/745 et Fabricants : 15% des fabricants seront prêts, 20% des dispositifs retirés du marché
Mise à jour de la FAQ du MDCG pour les organismes notifiés
Profils des fabricants de dispositifs médicaux : nombre d’employés, chiffre d’affaire, cœur de métier, classe des dispositifs, pays …
Le document du COCIR qui mettant en regard exigences de la directive machine et exigences du RDM.

Guide relatif à l’application des dispositions transitoires concernant la validité des certificats délivrés conformément aux directives 90/385/CEE et 93/42/CEE
Guide de l’IMDRF pour la cybersécurité des dispositifs médicaux
Le formulaire MIR passe en 7.2
formulaire pour répondre à l’appel à manifestation d’intérêt pour les groupes d’experts sur les dispositifs médicaux
Définition de dispositif wellness/bien-être selon la FDA
Notice de la HPRA relative à l’impact du Brexit sur le secteur des dispositifs médicaux et les risques de pénurie associés
Modèle de dossier d’investigation clinique (pour soumission à un comité d’éthique), conformément aux exigences du règlement UE 2017/745 (annexe XIV).
Ce document fait une bonne synthèse des exigences du RDM tout en évoquant explicitement les normes applicables.
Publication d’un appel à manifestation d’intérêt en vue de la constitution de groupes d’experts européens sur les dispositifs médicaux et DM-DIV

Enquête auprès des fabricants de dispositifs médicaux, par rapport à l’impact du nouveau règlement 2017/745
Guide du MDCG relatif à la déclaration des incidents liés aux implants mamaires
Guide du MDCG relatif à la déclaration des incidents pour les DM implantables actifs (pacemakers, sondes, défibrilateurs, …)
Nouveau format DICOM-RTV pour les applications vidéos médicales
La MHRA a mis à jour ses consignes en vue du hard Brexit, un guide détaille le rôle de la personne responsable, un rôle rendu.
Résumé du guide du MDCG pour le RCSPC

Synthèse des exigences et des bonnes pratiques en matière de Résumé des Caractéristiques de Sécurité et des Performances Cliniques pour les dispositifs médicaux
TÜV Rheinland devient le cinquième organisme à être notifié pour le règlement (ue) 2017/745
Medtech Europe a publié une FAQ sur le rôle des opérateurs économiques dans les règlements DM et DMDIV Résumé 1. Le fabricant ou l’importateur devrait-il.
La décision d’exécution 2019/1396 a été publiée le 10 septembre 2019, elle précise les critères pour la désignation de groupes d’experts dans le domaine des.
L’exécutif européen a changé de têtes, Ursula von der Leyen devient, depuis juillet, la présidente de la commission européenne et Stella Kyriakides, psychologue clinicienne chypriote,.
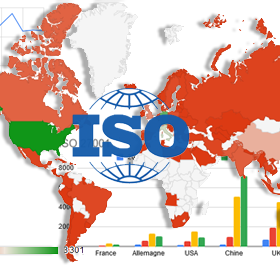
L’ISO a mis à jour son observatoire de l’utilisation des normes “système qualité”, l’occasion de voir les évolutions pour l’ISO 13485, la norme SMQ pour.

La certification ISO 13485 est-elle obligatoire pour le médical ?
[article initialement publié le 20 mars 2019, mis à jour le 10 septembre 2019] Le SCHEER vient de publier la version finale de ses lignes.
Nouveau document de travail de la FDA, dédié aux dispositifs destinés à la perte de poids il fournit néanmoins des idées généralisables sur l’évaluation du.
La FDA a publié le 30 août 2019 le guide “Consideration of Uncertainty in Making Benefit-Risk Determinations in Medical Device Premarket Approvals, De Novo Classifications,.